 |
Rx only
AZACTAM (aztreonam for injection, USP) contains the active ingredient, aztreonam, a monobactam. It was originally isolated from Chromobacterium violaceum . It is a synthetic bactericidal antibiotic.
The monobactams, having a unique monocyclic beta-lactam nucleus, are structurally different from other beta-lactam antibiotics (e.g., penicillins, cephalosporins, cephamycins). The sulfonic acid substituent in the 1-position of the ring activates the beta-lactam moiety; an aminothiazolyl oxime side chain in the 3-position and a methyl group in the 4-position confer the specific antibacterial spectrum and beta-lactamase stability.
Aztreonam is designated chemically as (Z)-2-[[[(2-amino-4-thiazolyl)[[(2S,-3S)-2-methyl-4-oxo-1-sulfo-3- azetidinyl]carbamoyl]methylene]amino]oxy]-2-methylpropionic acid.
AZACTAM is a sterile, nonpyrogenic, sodium-free, white to yellowish-white lyophilized cake containing approximately 780 mg arginine per gram of aztreonam. Following constitution, the product is for intramuscular or intravenous use. Aqueous solutions of the product have a pH in the range of 4.5 to 7.5.
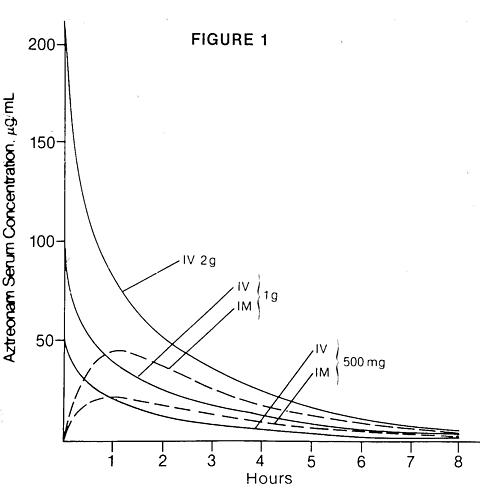
Single 30-minute intravenous infusions of 500 mg, 1 g and 2 g doses of AZACTAM (aztreonam for injection, USP) in healthy subjects produced aztreonam peak serum levels of 54, 90 and 204 µg/mL, respectively, immediately after administration; at eight hours, serum levels were 1, 3 and 6 µg/mL, respectively (Figure 1). Single 3-minute intravenous injections of the same doses resulted in serum levels of 58, 125 and 242 µg/mL at five minutes following completion of injection.
Serum concentrations of aztreonam in healthy subjects following completion of single intramuscular injections of 500 mg and 1 g doses are depicted in Figure 1; maximum serum concentrations occur at about one hour. After identical single intravenous or intramuscular doses of AZACTAM, the serum concentrations of aztreonam are comparable at one hour (1.5 hours from start of intravenous infusion) with similar slopes of serum concentrations thereafter.
|
|
The serum levels of aztreonam following single 500 mg or 1 g (intramuscular or intravenous) or 2 g (intravenous) doses of AZACTAM exceed the MIC 90 for Neisseria sp., H. influenzae and most genera of the Enterobacteriaceae for eight hours (for Enterobacter sp., the eight-hour serum levels exceed the MIC for 80 percent of strains). For Ps. aeruginosa, a single 2-g intravenous dose produces serum levels that exceed the MIC 90 for approximately four to six hours. All of the above doses of AZACTAM result in average urine levels of aztreonam that exceed the MIC 90 for the same pathogens for up to 12 hours.
When aztreonam pharmacokinetics were assessed for adult and pediatric patients, they were found to be comparable (down to 9 months old). The serum half-life of aztreonam averaged 1.7 hours (1.5 to 2.0) in subjects with normal renal function, independent of the dose and route of administration. In healthy subjects, based on a 70 kg person, the serum clearance was 91 mL/min and renal clearance was 56 mL/min; the apparent mean volume of distribution at steady-state averaged 12.6 liters, approximately equivalent to extracellular fluid volume.
In a study of healthy elderly male subjects (65 to 75 years of age), the average elimination half-life of aztreonam was slightly longer than in young healthy males.
In patients with impaired renal function, the serum half-life of aztreonam is prolonged (see DOSAGE AND ADMINISTRATION , Renal Impairment in Adult Patients ). The serum half-life of aztreonam is only slightly prolonged in patients with hepatic impairment since the liver is a minor pathway of excretion.
Average urine concentrations of aztreonam were approximately 1100, 3500 and 6600 µg/mL within the first two hours following single 500 mg, 1-g and 2-g intravenous doses of AZACTAM (30-minute infusions), respectively. The range of average concentrations for aztreonam in the 8- to 12-hour urine specimens in these studies was 25 to 120 µg/mL. After intramuscular injection of single 500 mg and 1 g doses of AZACTAM (aztreonam for injection, USP), urinary levels were approximately 500 and 1200 µg/mL, respectively, within the first two hours, declining to 180 and 470 µg/mL in the six to eight hour specimens. In healthy subjects, aztreonam is excreted in the urine about equally by active tubular secretion and glomerular filtration. Approximately 60 to 70 percent of an intravenous or intramuscular dose was recovered in the urine by eight hours. Urinary excretion of a single parenteral dose was essentially complete by 12 hours after injection. About 12 percent of a single intravenous radiolabeled dose was recovered in the feces. Unchanged aztreonam and the inactive beta-lactam ring hydrolysis product of aztreonam were present in feces and urine.
Intravenous or intramuscular administration of a single 500 mg or 1 g dose of AZACTAM every eight hours for seven days to healthy subjects produced no apparent accumulation of aztreonam or modification of its disposition characteristics; serum protein binding averaged 56 percent and was independent of dose. An average of about 6 percent of a 1 g intramuscular dose was excreted as a microbiologically inactive open beta-lactam ring hydrolysis product (serum half-life approximately 26 hours) of aztreonam in the zero to eight hour urine collection on the last day of multiple dosing.
Renal function was monitored in healthy subjects given aztreonam; standard tests (serum creatinine, creatinine clearance, BUN, urinalysis and total urinary protein excretion) as well as special tests (excretion of N-acetyl-(beta)-glucosaminidase, alanine aminopeptidase and (beta) 2 -microglobulin) were used. No abnormal results were obtained.
Aztreonam achieves measurable concentrations in the following body fluids and tissues:
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The concentration of aztreonam in saliva at 30 minutes after a single 1 g intravenous dose (9 patients) was 0.2 µg/mL; in human milk at two hours after a single 1 g intravenous dose (6 patients), 0.2 µg/mL, and at six hours after a single 1 g intramuscular dose (6 patients), 0.3 µg/mL; in amniotic fluid at six to eight hours after a single 1 g intravenous dose (5 patients), 2 µg/mL. The concentration of aztreonam in peritoneal fluid obtained one to six hours after multiple 2 g intravenous doses ranged between 12 and 90 µg/mL in 7 of 8 patients studied.
Aztreonam given intravenously rapidly reaches therapeutic concentrations in peritoneal dialysis fluid; conversely, aztreonam given intraperitoneally in dialysis fluid rapidly produces therapeutic serum levels.
Concomitant administration of probenecid or furosemide and AZACTAM (aztreonam for injection, USP) causes clinically insignificant increases in the serum levels of aztreonam. Single-dose intravenous pharmacokinetic studies have not shown any significant interaction between aztreonam and concomitantly administered gentamicin, nafcillin sodium, cephradine, clindamycin or metronidazole. No reports of disulfiram-like reactions with alcohol ingestion have been noted; this is not unexpected since aztreonam does not contain a methyl-tetrazole side chain.
Aztreonam exhibits potent and specific activity in vitro against a wide spectrum of gram-negative aerobic pathogens including Pseudomonas aeruginosa . The bactericidal action of aztreonam results from the inhibition of bacterial cell wall synthesis due to a high affinity of aztreonam for penicillin binding protein 3 (PBP3). Aztreonam, unlike the majority of beta-lactam antibiotics, does not induce beta-lactamase activity and its molecular structure confers a high degree of resistance to hydrolysis by beta-lactamases (i.e., penicillinases and cephalosporinases) produced by most gram-negative and gram-positive pathogens; it is, therefore, usually active against gram-negative aerobic microorganisms that are resistant to antibiotics hydrolyzed by beta-lactamases. It is active against many strains that are multiply-resistant to other antibiotics, such as certain cephalosporins, penicillin, and aminoglycosides. Aztreonam maintains its antimicrobial activity over a pH range of 6 to 8 in vitro , as well as in the presence of human serum and under anaerobic conditions.
Aztreonam has been shown to be active against most strains of the following microorganisms, both in vitro and in clinical infections as described in the section
Aerobic gram-negative microorganisms:
Citrobacter species, including C. freundii
Enterobacter species, including E. cloacae
Escherichia coli
Haemophilus influenzae (including ampicillin-resistant and other penicillinase-producing strains)
Klebsiella oxytoca
Klebsiella pneumoniae
Proteus mirabilis
Pseudomonas aeruginosa
Serratia species, including S. marcescens
The following in vitro data are available, but their clinical significance is unknown .
Aztreonam exhibits in vitro minimal inhibitory concentrations (MIC's) of 8 µg/mL or less against most (>/=90%) strains of the following microorganisms; however, the safety and effectiveness of aztreonam in treating clinical infections due to these microorganisms have not been established in adequate and well-controlled clinical trials.
Aerobic gram-negative microorganisms:
Aeromonas hydrophila
Morganella morganii
Neisseria gonorrhoeae (including penicillinase-producing strains)
Pasteurella multocida
Proteus vulgaris
Providencia stuartii
Providencia rettgeri
Yersinia enterocolitica
Aztreonam and aminoglycosides have been shown to be synergistic in vitro against most strains of P. aeruginosa , many strains of Enterobacteriaceae , and other gram-negative aerobic bacilli.
Alterations of the anaerobic intestinal flora by broad spectrum antibiotics may decrease colonization resistance, thus permitting overgrowth of potential pathogens, e.g., Candida and clostridium species. Aztreonam has little effect on the anaerobic intestinal microflora in in vitro studies. Clostridium difficile and its cytotoxin were not found in animal models following administration of aztreonam. (See ADVERSE REACTIONS , Gastrointestinal .)
Dilution Techniques: Quantitative methods are used to determine antimicrobial minimal inhibitory concentrations (MIC's). These MIC's provide estimates of the susceptibility of bacteria to antimicrobial compounds. The MIC's should be determined using a standardized procedure. Standardized procedures are based on a dilution method 1 (broth or agar) or equivalent with standardized inoculum concentrations and standardized concentrations of aztreonam powder. The MIC values should be interpreted according to the following criteria:
For testing aerobic microorganisms other than Haemophilus influenzae:
|
When testing Haemophilus influenzae a :
|
||||||||
A report of "Susceptible" indicates that the pathogen is likely to be inhibited if the antimicrobial compound in the blood reaches the concentrations usually achievable. A report of "Intermediate" indicates that the result should be considered equivocal, and, if the microorganism is not fully susceptible to alternative, clinically feasible drugs, the test should be repeated. This category implies possible clinical applicability in body sites where the drug is physiologically concentrated or in situations where high dosage of drug can be used. This category also provides a buffer zone which prevents small uncontrolled technical factors from causing major discrepancies in interpretation. A report of "Resistant" indicates that the pathogen is not likely to be inhibited if the antimicrobial compound in the blood reaches the concentrations usually achievable; other therapy should be selected.
Standardized susceptibility test procedures require the use of laboratory control microorganisms to control the technical aspects of the laboratory procedures. Standard aztreonam powder should provide the following MIC values:
|
||||||||||
Diffusion Techniques: Quantitative methods that require measurement of zone diameters also provide reproducible estimates of the susceptibility of bacteria to antimicrobial compounds. One such standardized procedure 2 requires the use of standardized inoculum concentrations. This procedure uses paper disks impregnated with 30-µg aztreonam to test the susceptibility of microorganisms to aztreonam.
Reports from the laboratory providing results of the standard single-disk susceptibility test with a 30-µg aztreonam disk should be interpreted according to the following criteria:
For testing aerobic microorganisms other than Haemophilus influenzae:
|
When testing Haemophilus influenzae a :
|
||||||||
Interpretation should be as stated above for results using dilution techniques. Interpretation involves correlation of the diameter obtained in the disk test with the MIC for aztreonam.
As with standardized dilution techniques, diffusion methods require the use of laboratory control microorganisms that are used to control the technical aspects of the laboratory procedures. For the diffusion technique, the 30-µg aztreonam disk should provide the following zone diameters in these laboratory test quality control strains.
|
||||||||||
Before initiating treatment with AZACTAM, appropriate specimens should be obtained for isolation of the causative organism(s) and for determination of susceptibility to aztreonam. Treatment with AZACTAM may be started empirically before results of the susceptibility testing are available; subsequently, appropriate antibiotic therapy should be continued.
AZACTAM (aztreonam for injection, USP) is indicated for the treatment of the following infections caused by susceptible gram-negative microorganisms:
Urinary Tract Infections (complicated and uncomplicated), including pyelonephritis and cystitis (initial and recurrent) caused by Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis, Pseudomonas aeruginosa, Enterobacter cloacae, Klebsiella oxytoca * , Citrobacter species * and Serratia marcescens * .
Lower Respiratory Tract Infections, including pneumonia and bronchitis caused by Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Haemophilus influenzae, Proteus mirabilis, Enterobacter species and Serratia marcescens * .
Septicemia caused by Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Proteus mirabilis * , Serratia marcescens * and Enterobacter species
Skin and Skin-Structure Infections, including those associated with postoperative wounds, ulcers and burns caused by Escherichia coli, Proteus mirabilis, Serratia marcescens, Enterobacter species Pseudomonas aeruginosa, Klebsiella pneumoniae and Citrobacter species * .
Intra-abdominal Infections, including peritonitis caused by Escherichia coli, Klebsiella species including K. pneumoniae, Enterobacter species including E. cloacae * , Pseudomonas aeruginosa, Citrobacter species * including C. freundii * and Serratia species * including S. marcescens * .
Gynecologic Infections, including endometritis and pelvic cellulitis caused by Escherichia coli, Klebsiella pneumoniae * , Enterobacter species * including E. cloacae * and Proteus mirabilis * .
* Efficacy for this organism in this organ system was studied in fewer than ten infections.AZACTAM is indicated for adjunctive therapy to surgery in the management of infections caused by susceptible organisms, including abscesses, infections complicating hollow viscus perforations, cutaneous infections and infections of serous surfaces. AZACTAM is effective against most of the commonly encountered gram-negative aerobic pathogens seen in general surgery.
Concurrent initial therapy with other antimicrobial agents and AZACTAM (aztreonam for injection, USP) is recommended before the causative organism(s) is known in seriously ill patients who are also at risk of having an infection due to gram-positive aerobic pathogens. If anaerobic organisms are also suspected as etiologic agents, therapy should be initiated using an anti-anaerobic agent concurrently with AZACTAM (see DOSAGE AND ADMINISTRATION ). Certain antibiotics (e.g., cefoxitin, imipenem) may induce high levels of beta-lactamase in vitro in some gram-negative aerobes such as Enterobacter and Pseudomonas species, resulting in antagonism to many beta-lactam antibiotics including aztreonam. These in vitro findings suggest that such beta-lactamase inducing antibiotics not be used concurrently with aztreonam. Following identification and susceptibility testing of the causative organism(s), appropriate antibiotic therapy should be continued.
This preparation is contraindicated in patients with known hypersensitivity to aztreonam or any other component in the formulation.
Both animal and human data suggest that AZACTAM is rarely cross-reactive with other beta-lactam antibiotics and weakly immunogenic. Treatment with aztreonam can result in hypersensitivity reactions in patients with or without prior exposure. (See CONTRAINDICATIONS .)
Careful inquiry should be made to determine whether the patient has any history of hypersensitivity reactions to any allergens.
While cross-reactivity of aztreonam with other beta-lactam antibiotics is rare, this drug should be administered with caution to any patient with a history of hypersensitivity to beta-lactams (e.g., penicillins, cephalosporins, and/or carbapenems). Treatment with aztreonam can result in hypersensitivity reactions in patients with or without prior exposure to aztreonam. If an allergic reaction to aztreonam occurs, discontinue the drug and institute supportive treatment as appropriate (e.g., maintenance of ventilation, pressor amines, antihistamines, corticosteroids). Serious hypersensitivity reactions may require epinephrine and other emergency measures. (See ADVERSE REACTIONS .)
Pseudomembranous colitis has been reported with nearly all antibacterial agents, including aztreonam, and may range in severity from mild to life-threatening. Therefore, it is important to consider this diagnosis in patients who present with diarrhea subsequent to the administration of antibacterial agents.
Treatment with antibacterial agents alters the normal flora of the colon and may permit overgrowth of Clostridia. Studies indicate that a toxin produced by Clostridium difficile is one primary cause of "antibiotic-associated colitis."
After the diagnosis of pseudomembranous colitis has been established, therapeutic measures should be initiated. Mild cases of pseudomembranous colitis usually respond to drug discontinuation alone. In moderate to severe cases, consideration should be given to management with fluids and electrolytes, protein supplementation, and treatment with an antibacterial drug clinically effective against C. difficile colitis
Rare cases of toxic epidermal necrolysis have been reported in association with aztreonam in patients undergoing bone marrow transplant with multiple risk factors including sepsis, radiation therapy and other concomitantly administered drugs associated with toxic epidermal necrolysis.
In patients with impaired hepatic or renal function, appropriate monitoring is recommended during therapy.
If an aminoglycoside is used concurrently with aztreonam, especially if high dosages of the former are used or if therapy is prolonged, renal function should be monitored because of the potential nephrotoxicity and ototoxicity of aminoglycoside antibiotics.
The use of antibiotics may promote the overgrowth of nonsusceptible organisms, including gram-positive organisms ( Staphylococcus aureus and Streptococcus faecalis ) and fungi. Should superinfection occur during therapy, appropriate measures should be taken.
Carcinogenicity studies in animals have not been performed.
Genetic toxicology studies performed in vivo and in vitro with aztreonam in several standard laboratory models revealed no evidence of mutagenic potential at the chromosomal or gene level.
Two-generation reproduction studies in rats at daily doses up to 20 times the maximum recommended human dose, prior to and during gestation and lactation, revealed no evidence of impaired fertility. There was a slightly reduced survival rate during the lactation period in the offspring of rats that received the highest dosage, but not in offspring of rats that received five times the maximum recommended human dose.
Aztreonam crosses the placenta and enters the fetal circulation.
Studies in pregnant rats and rabbits, with daily doses up to 15 and 5 times, respectively, the maximum recommended human dose, revealed no evidence of embryo- or fetotoxicity or teratogenicity. No drug induced changes were seen in any of the maternal, fetal, or neonatal parameters that were monitored in rats receiving 15 times the maximum recommended human dose of aztreonam during late gestation and lactation.
There are no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, aztreonam should be used during pregnancy only if clearly needed.
Aztreonam is excreted in breast milk in concentrations that are less than 1 percent of concentrations determined in simultaneously obtained maternal serum; consideration should be given to temporary discontinuation of nursing and use of formula feedings.
The safety and effectiveness of intravenous AZACTAM (aztreonam for injection, USP) have been established in the age groups 9 months to 16 years. Use of AZACTAM in these age groups is supported by evidence from adequate and well-controlled studies of AZACTAM in adults with additional efficacy, safety, and pharmacokinetic data from non-comparative clinical studies in pediatric patients. Sufficient data are not available for pediatric patients under 9 months of age or for the following treatment indications/pathogens: septicemia and skin and skin-structure infections (where the skin infection is believed or known to be due to H. influenzae type b). In pediatric patients with cystic fibrosis, higher doses of AZACTAM may be warranted. (See , DOSAGE AND ADMINISTRATION , and CLINICAL STUDIES .)
Local reactions such as phlebitis/thrombophlebitis following IV administration, and discomfort/swelling at the injection site following IM administration occurred at rates of approximately 1.9 percent and 2.4 percent, respectively.
Systemic reactions (considered to be related to therapy or of uncertain etiology) occurring at an incidence of 1 to 1.3 percent include diarrhea, nausea and/or vomiting, and rash. Reactions occurring at an incidence of less than 1 percent are listed within each body system in order of decreasing severity:
Hypersensitivity-- anaphylaxis, angioedema, bronchospasm.
Hematologic-- pancytopenia, neutropenia, thrombocytopenia, anemia, eosinophilia, leukocytosis, thrombocytosis.
Gastrointestinal abdominal cramps; rare cases of C. difficile -associated diarrhea, including pseudomembraneous colitis, or gastrointestinal bleeding have been reported. Onset of pseudomembranous colitis symptoms may occur during or after antibiotic treatment. (See .)
Dermatologic --toxic epidermal necrolysis (see ), purpura, erythema multiforme, exfoliative dermatitis, urticaria, petechiae, pruritus, diaphoresis.
Cardiovascular --hypotension, transient ECG changes (ventricular bigeminy and PVC), flushing.
Respiratory --wheezing, dyspnea, chest pain.
Hepatobiliary --hepatitis, jaundice.
Nervous System --seizure, confusion, vertigo, paresthesia, insomnia, dizziness.
Musculoskeletal --muscular aches.
Special Senses --tinnitus, diplopia, mouth ulcer, altered taste, numb tongue, sneezing, nasal congestion, halitosis.
Other --vaginal candidiasis, vaginitis, breast tenderness.
Body as a Whole --weakness, headache, fever, malaise.
Of the 612 pediatric patients who were treated with AZACTAM in clinical trials, less than 1% required discontinuation of therapy due to adverse events. The following systemic adverse events, regardless of drug relationship, occurred in at least 1% of treated patients in domestic clinical trials: rash (4.3%), diarrhea (1.4%), and fever (1.0%). These adverse events were comparable to those observed in adult clinical trials.
In 343 pediatric patients receiving intravenous therapy, the following local reactions were noted: pain (12%), erythema (2.9%), induration (0.9%), and phlebitis (2.1%). In the US patient population, pain occurred in 1.5% of patients, while each of the remaining three local reactions had an incidence of 0.5%.
The following laboratory adverse events, regardless of drug relationship, occurred in at least 1% of treated patients: increased eosinophils (6.3%), increased platelets (3.6%), neutropenia (3.2%), increased AST (3.8%), increased ALT (6.5%), and increased serum creatinine (5.8%).
In US pediatric clinical trials, neutropenia (absolute neutrophil count less than 1000/mm 3 ) occurred in 11.3% of patients (8/71) younger than 2 years receiving 30 mg/kg q6h. AST and ALT elevations to greater than 3 times the upper limit of normal were noted in 15-20% of patients aged 2 years or above receiving 50 mg/kg q6h. The increased frequency of these reported laboratory adverse events may be due to either increased severity of illness treated or higher doses of AZACTAM (aztreonam for injection, USP) administered.
Adverse laboratory changes without regard to drug relationship that were reported during clinical trials were:
Hepatic-- elevations of AST (SGOT), ALT (SGPT), and alkaline phosphatase; signs or symptoms of hepatobiliary dysfunction occurred in less than 1 percent of recipients (see above).
Hematologic-- increases in prothrombin and partial thromboplastin times, positive Coombs test.
Renal increases in serum creatinine.
If necessary, aztreonam may be cleared from the serum by hemodialysis and/or peritoneal dialysis.
AZACTAM may be administered intravenously or by intramuscular injection. Dosage and route of administration should be determined by susceptibility of the causative organisms, severity and site of infection, and the condition of the patient.
The intravenous route is recommended for patients requiring single doses greater than 1 g or those with bacterial septicemia, localized parenchymal abscess (e.g., intra-abdominal abscess), peritonitis or other severe systemic or life-threatening infections.
The duration of therapy depends on the severity of infection. Generally, AZACTAM should be continued for at least 48 hours after the patient becomes asymptomatic or evidence of bacterial eradication has been obtained. Persistent infections may require treatment for several weeks. Doses smaller than those indicated should not be used.
Prolonged serum levels of aztreonam may occur in patients with transient or persistent renal insufficiency. Therefore, the dosage of AZACTAM should be halved in patients with estimated creatinine clearances between 10 and 30 mL/min/1.73 m 2 after an initial loading dose of 1 g or 2 g.
When only the serum creatinine concentration is available, the following formula (based on sex, weight, and age of the patient) may be used to approximate the creatinine clearance (Clcr). The serum creatinine should represent a steady state of renal function.
| Males: Clcr = |
weight (kg) × (140-age)
72 × serum creatinine (mg/dL) |
| Females: 0.85 × above value | |
In patients with severe renal failure (creatinine clearance less than 10 mL/min/1.73 m 2 ), such as those supported by hemodialysis, the usual dose of 500 mg, 1 g or 2 g should be given initially. The maintenance dose should be one-fourth of the usual initial dose given at the usual fixed interval of 6, 8 or 12 hours. For serious or life-threatening infections, in addition to the maintenance doses, one-eighth of the initial dose should be given after each hemodialysis session.
Renal status is a major determinant of dosage in the elderly; these patients in particular may have diminished renal function. Serum creatinine may not be an accurate determinant of renal status. Therefore, as with all antibiotics eliminated by the kidneys, estimates of creatinine clearance should be obtained, and appropriate dosage modifications made if necessary.
AZACTAM (aztreonam for injection, USP) should be administered intravenously to pediatric patients with normal renal function. There are insufficient data regarding intramuscular administration to pediatric patients or dosing in pediatric patients with renal impairment. (See PRECAUTIONS : Pediatric Use .)
|
||||||||||||||||||||||||||||||
Because of the serious nature of infections due to Pseudomonas aeruginosa , dosage of 2 g every six or eight hours is recommended, at least upon initiation of therapy, in systemic infections caused by this organism in adults.
A total of 612 pediatric patients aged 1 month to 12 years were enrolled in uncontrolled clinical trials of aztreonam in the treatment of serious gram-negative infections, including urinary tract, lower respiratory tract, skin and skin-structure, and intra-abdominal infections.
Upon the addition of the diluent to the container, contents should be shaken immediately and vigorously . Constituted solutions are not for multiple-dose use; should the entire volume in the container not be used for a single-dose, the unused solution must be discarded.
Depending upon the concentration of aztreonam and diluent used, constituted AZACTAM yields a colorless to light straw yellow solution which may develop a slight pink tint on standing (potency is not affected). Parenteral drug products should be inspected visually for particulate matter and discoloration whenever solution and container permit.
Intravenous infusion solutions of AZACTAM not exceeding 2% w/v prepared with Sodium Chloride Injection USP 0.9% or Dextrose Injection USP 5%, to which clindamycin phosphate, gentamicin sulfate, tobramycin sulfate, or cefazolin sodium have been added at concentrations usually used clinically, are stable for up to 48 hours at room temperature or seven days under refrigeration. Ampicillin sodium admixtures with aztreonam in Sodium Chloride Injection USP 0.9% are stable for 24 hours at room temperature and 48 hours under refrigeration; stability in Dextrose Injection USP 5% is two hours at room temperature and eight hours under refrigeration.
Aztreonam-cloxacillin sodium and aztreonam-vancomycin hydrochloride admixtures are stable in Dianeal® 137 (Peritoneal Dialysis Solution) with 4.25% Dextrose for up to 24 hours at room temperature.
Aztreonam is incompatible with nafcillin sodium, cephradine, and metronidazole.
Other admixtures are not recommended since compatibility data are not available.
For Bolus Injection : The contents of an AZACTAM (aztreonam for injection, USP) 15 mL or 30 mL capacity vial should be constituted with 6 to 10 mL Sterile Water for Injection USP. For Infusion: Contents of the 100 mL capacity bottle should be constituted to a final concentration not exceeding 2% w/v (at least 50 mL of any appropriate infusion solution listed below per gram aztreonam). These solutions may be frozen immediately after constitution in the original container. (See Stability below.)
If the contents of a 15 mL or 30 mL capacity vial are to be transferred to an appropriate infusion solution, each gram of aztreonam should be initially constituted with at least 3 mL Sterile Water for Injection USP. Further dilution may be obtained with one of the following intravenous infusion solutions:
Sodium Chloride Injection USP, 0.9%
Ringer's Injection USP
Lactated Ringer' Injection USP
Dextrose Injection USP, 5% or 10%
Dextrose and Sodium Chloride Injection USP, 5%:0.9%,
5%:0.45% or 5%:0.2%
Sodium Lactate Injection USP (M/6 Sodium Lactate)
Ionosol® B and 5% Dextrose
Isolyte® E
Isolyte® E with 5% Dextrose
Isolyte® M with 5% Dextrose
Normosol®-R
Normosol®-R and 5% Dextrose
Normosol®-M and 5% Dextrose
Mannitol Injection USP, 5% or 10%
Lactated Ringer' and 5% Dextrose Injection
Plasma-Lyte® M and 5% Dextrose
10% Travert® Injection
10% Travert® and Electrolyte No. 1 Injection
10% Travert® and Electrolyte No. 2 Injection
10% Travert® and Electrolyte No. 3 Injection
The contents of an AZACTAM 15 mL or 30 mL capacity vial should be constituted with at least 3 mL of an appropriate diluent per gram aztreonam. The following diluents may be used:
Sterile Water for Injection USP
Sterile Bacteriostatic Water for Injection, USP (with benzyl alcohol or with methyl- and propylparabens)
Sodium Chloride Injection USP, 0.9%
Bacteriostatic Sodium Chloride Injection USP (with benzyl alcohol)
AZACTAM solutions for IV infusion at concentrations not exceeding 2% w/v must be used within 48 hours following constitution if kept at controlled room temperature (59°- 86° F/15°- 30° C) or within seven days if refrigerated (36°- 46° F/ 2°- 8° C).
Frozen aztreonam infusion solutions may be stored for up to three months at -4° F/-20° C; frozen solutions may be thawed at controlled room temperature or by overnight refrigeration. Solutions that have been thawed and maintained at controlled room temperature or under refrigeration should be used within 24 or 72 hours after removal from the freezer, respectively. Solutions should not be refrozen.
AZACTAM solutions at concentrations exceeding 2% w/v, except those prepared with Sterile Water for Injection USP or Sodium Chloride Injection USP, should be used promptly after preparation; the two excepted solutions must be used within 48 hours if stored at controlled room temperature or within seven days if refrigerated.
Bolus Injection: A bolus injection may be used to initiate therapy. The dose should be slowly injected directly into a vein, or the tubing of a suitable administration set, over a period of three to five minutes (see next paragraph regarding flushing of tubing).
Infusion: With any intermittent infusion of aztreonam and another drug with which it is not pharmaceutically compatible, the common delivery tube should be flushed before and after delivery of aztreonam with any appropriate infusion solution compatible with both drug solutions; the drugs should not be delivered simultaneously. Any AZACTAM (aztreonam for injection, USP) infusion should be completed within a 20 to 60 minute period. With use of a Y-type administration set , careful attention should be given to the calculated volume of aztreonam solution required so that the entire dose will be infused. A volume control administration set may be used to deliver an initial dilution of AZACTAM (see Preparation Of Parenteral Solutions, For Infusion ) into a compatible infusion solution during administration; in this case, the final dilution of aztreonam should provide a concentration not exceeding 2% w/v.
The dose should be given by deep injection into a large muscle mass (such as the upper outer quadrant of the gluteus maximus or lateral part of the thigh). Aztreonam is well tolerated and should not be admixed with any local anesthetic agent.
AZACTAM® (aztreonam for injection, USP)-Lyophilized
Single-dose 15 mL capacity vials:
500 mg/vial: Packages of 10 (NDC 51479-050-05)
1 g/vial: Packages of 10 (NDC 51479-051-15)
Single-dose 30 mL capacity vial:
2 g/vial: Packages of 10 (NDC 51479-052-30)
Single-dose 100 mL capacity intravenous infusion bottles with bail bands:
1 g/bottle: Packages of 10 (NDC 51479-051-10)
2 g/bottle: Packages of 10 (NDC 51479-052-10)
Store original packages at room temperature; avoid excessive heat.
AZACTAM® (aztreonam injection) in Galaxy® plastic container (PL 2040) as a frozen, 50 mL single-dose intravenous solution as follows:
1 g aztreonam/50 mL container: Packages of 24 (NDC 51479-048-01)
2 g aztreonam/50 mL container: Packages of 24 (NDC 51479-049-01)
AZACTAM® is a registered trademark of Bristol-Myers Squibb Company
Manufactured by
Bristol-Myers Squibb Company
Princeton, NJ 08543 U.S.A.
Distributed by
DURA Pharmaceuticals, Inc.
San Diego, CA 92121 U.S.A.
Revised March 1999
J4-671
AZL001A99
 |