 |
|
Dantrium (dantrolene sodium) has a potential for hepatotoxicity, and should not be used in conditions other than those recommended. Symptomatic hepatitis (fatal and non-fatal) has been reported at various dose levels of the drug. The incidence reported in patients taking up to 400 mg/day is much lower than in those taking doses of 800 mg or more per day. Even sporadic short courses of these higher dose levels within a treatment regimen markedly increased the risk of serious hepatic injury. Liver dysfunction as evidenced by blood chemical abnormalities alone (liver enzyme elevations) has been observed in patients exposed to Dantrium for varying periods of time. Overt hepatitis has occurred at varying intervals after initiation of therapy, but has been most frequently observed between the third and twelfth month of therapy. The risk of hepatic injury appears to be greater in females, in patients over 35 years of age, and in patients taking other medication(s) in addition to Dantrium (dantrolene sodium). Dantrium should be used only in conjunction with appropriate monitoring of hepatic function including frequent determination of SGOT or SGPT. If no observable benefit is derived from the administration of Dantrium after a total of 45 days, therapy should be discontinued. The lowest possible effective dose for the individual patient should be prescribed. |
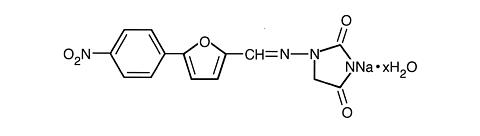
The chemical formula of Dantrium (dantrolene sodium) is hydrated 1-[[[5-(4-nitrophenyl)-2-furanyl]methylene]amino]-2, 4-imidazolidinedione sodium salt. It is an orange powder, slightly soluble in water, but due to its slightly acidic nature the solubility increases somewhat in alkaline solution. The anhydrous salt has a molecular weight of 336. The hydrated salt contains approximately 15% water (3-1/2 moles) and has a molecular weight of 399. The structural formula for the hydrated salt is:
|
|
Dantrium is supplied in capsules
of 25 mg, 50 mg, and 100 mg.
Inactive Ingredients: Each capsule contains edible black ink, FD&C Yellow No. 6, gelatin, lactose, magnesium stearate, starch, synthetic iron oxide red, synthetic iron oxide yellow, talc, and titanium dioxide.
In isolated nerve-muscle preparation, Dantrium has been shown to produce relaxation by affecting the contractile response of the skeletal muscle at a site beyond the myoneural junction, directly on the muscle itself. In skeletal muscle, Dantrium dissociates the excitation-contraction coupling, probably by interfering with the release of Ca++ from the sarcoplasmic reticulum. This effect appears to be more pronounced in fast muscle fibers as compared to slow ones, but generally affects both. A central nervous system effect occurs, with drowsiness, dizziness, and generalized weakness occasionally present. Although Dantrium does not appear to directly affect the CNS, the extent of its indirect effect is unknown. The absorption of Dantrium after oral administration in humans is incomplete and slow but consistent, and dose-related blood levels are obtained. The duration and intensity of skeletal muscle relaxation is related to the dosage and blood levels. The mean biologic half-life of Dantrium in adults is 8.7 hours after a 100-mg dose. Specific metabolic pathways in the degradation and elimination of Dantrium in human subjects have been established. Metabolic patterns are similar in adults and pediatric patients. In addition to the parent compound, dantrolene, which is found in measurable amounts in blood and urine, the major metabolites noted in body fluids are the 5-hydroxy analog and the acetamido analog. Since Dantrium is probably metabolized by hepatic microsomal enzymes, enhancement of its metabolism by other drugs is possible. However, neither phenobarbital nor diazepam appears to affect Dantrium metabolism
Clinical experience in the management of fulminant human malignant hyperthermia, as well as experiments conducted in malignant hyperthermia susceptible swine, have revealed that the administration of intravenous dantrolene, combined with indicated supportive measures, is effective in reversing the hypermetabolic process of malignant hyperthermia. Known differences between human and swine malignant hyperthermia are minor. The prophylactic administration of oral or intravenous dantrolene to malignant hyperthermia susceptible swine will attenuate or prevent the development of signs of malignant hyperthermia in a manner dependent upon the dosage of dantrolene administered and the intensity of the malignant hyperthermia triggering stimulus. Limited clinical experience with the administration of oral dantrolene to patients judged malignant hyperthermia susceptible, when combined with clinical experience in the use of intravenous dantrolene for the treatment of malignant hyperthermia and data derived from the above cited animal model experiments, suggests that oral dantrolene will also attenuate or prevent the development of signs of human malignant hyperthermia, provided that currently accepted practices in the management of such patients are adhered to (see ); intravenous dantrolene should also be available for use should the signs of malignant hyperthermia appear.
Dantrium is indicated in controlling the manifestations of clinical spasticity resulting from upper motor neuron disorders (e.g., spinal cord injury, stroke, cerebral palsy, or multiple sclerosis). It is of particular benefit to the patient whose functional rehabilitation has been retarded by the sequelae of spasticity. Such patients must have presumably reversible spasticity where relief of spasticity will aid in restoring residual function. Dantrium is not indicated in the treatment of skeletal muscle spasm resulting from rheumatic disorders.
If improvement occurs, it will ordinarily occur within the dosage titration (see DOSAGE AND ADMINISTRATION ), and will be manifested by a decrease in the severity of spasticity and the ability to resume a daily function not quite attainable without Dantrium .
Occasionally, subtle but meaningful improvement in spasticity may occur with Dantrium therapy. In such instances, information regarding improvement should be solicited from the patient and those who are in constant daily contact and attendance with him. Brief withdrawal of Dantrium for a period of 2 to 4 days will frequently demonstrate exacerbation of the manifestations of spasticity and may serve to confirm a clinical impression.
A decision to continue the administration of Dantrium on a long-term basis is justified if introduction of the drug into the patient' regimen:
produces a significant reduction in painful and/or disabling spasticity such as clonus, or
permits a significant reduction in the intensity and/or degree of nursing care required, or
rids the patient of any annoying manifestation of spasticity considered important by the patient himself.
Oral Dantrium is also indicated preoperatively to prevent or attenuate the development of signs of malignant hyperthermia in known, or strongly suspect, malignant hyperthermia susceptible patients who require anesthesia and/or surgery. Currently accepted clinical practices in the management of such patients must still be adhered to (careful monitoring for early signs of malignant hyperthermia, minimizing exposure to triggering mechanisms and prompt use of intravenous dantrolene sodium and indicated supportive measures should signs of malignant hyperthermia appear); see also the package insert for Dantrium ® (dantrolene sodium) Intravenous .
Oral Dantrium should be administered following a malignant hyperthermic crisis to prevent recurrence of the signs of malignant hyperthermia.
Active hepatic disease, such as hepatitis and cirrhosis, is a contraindication for use of Dantrium . Dantrium is contraindicated where spasticity is utilized to sustain upright posture and balance in locomotion or whenever spasticity is utilized to obtain or maintain increased function.
It is important to recognize that fatal and non-fatal liver disorders of an idiosyncratic or hypersensitivity type may occur with Dantrium therapy
At the start of Dantrium therapy, it is desirable to do liver function studies (SGOT, SGPT, alkaline phosphatase, total bilirubin) for a baseline or to establish whether there is pre-existing liver disease. If baseline liver abnormalities exist and are confirmed, there is a clear possibility that the potential for Dantrium hepatotoxicity could be enhanced, although such a possibility has not yet been established.
Liver function studies (e.g., SGOT or SGPT) should be performed at appropriate intervals during Dantrium therapy. If such studies reveal abnormal values, therapy should generally be discontinued. Only where benefits of the drug have been of major importance to the patient, should reinitiation or continuation of therapy be considered. Some patients have revealed a return to normal laboratory values in the face of continued therapy while others have not.
If symptoms compatible with hepatitis, accompanied by abnormalities in liver function tests or jaundice appear, Dantrium should be discontinued. If caused by Dantrium and detected early, the abnormalities in liver function characteristically have reverted to normal when the drug was discontinued.
Dantrium therapy has been reinstituted in a few patients who have developed clinical and/or laboratory evidence of hepatocellular injury. If such reinstitution of therapy is done, it should be attempted only in patients who clearly need Dantrium and only after previous symptoms and laboratory abnormalities have cleared. The patient should be hospitalized and the drug should be restarted in very small and gradually increasing doses. Laboratory monitoring should be frequent and the drug should be withdrawn immediately if there is any indication of recurrent liver involvement. Some patients have reacted with unmistakable signs of liver abnormality upon administration of a challenge dose, while others have not.
Dantrium should be used with particular caution in females and in patients over 35 years of age in view of apparent greater likelihood of drug-induced, potentially fatal, hepatocellular disease in these groups.
Long-term safety of Dantrium in humans has not been established. Chronic studies in rats, dogs, and monkeys at dosages greater than 30 mg/kg/day showed growth or weight depression and signs of hepatopathy and possible occlusion nephropathy, all of which were reversible upon cessation of treatment. Sprague-Dawley female rats fed dantrolene sodium for 18 months at dosage levels of 15, 30, and 60 mg/kg/day showed an increased incidence of benign and malignant mammary tumors compared with concurrent controls. At the highest dose level, there was an increase in the incidence of benign hepatic lymphatic neoplasms. In a 30-month study at the same dose levels also in Sprague-Dawley rats, dantrolene sodium produced a decrease in the time of onset of mammary neoplasms. Female rats at the highest dose level showed an increased incidence of hepatic lymphangiomas and hepatic angiosarcomas.
The only drug-related effect seen in a 30-month study in Fischer-344 rats was a dose-related reduction in the time of onset of mammary and testicular tumors. A 24-month study in HaM/ICR mice revealed no evidence of carcinogenic activity. Carcinogenicity in humans cannot be fully excluded, so that this possible risk of chronic administration must be weighed against the benefits of the drug (i.e., after a brief trial) for the individual patient.
The safety of Dantrium for use in women who are or who may become pregnant has not been established. Dantrium should not be used in nursing mothers.
Usage in Pediatric Patients: The long-term safety of Dantrium in pediatric patients under the age of 5 years has not been established. Because of the possibility that adverse effects of the drug could become apparent only after many years, a benefit-risk consideration of the long-term use of Dantrium is particularly important in pediatric patients.
Drowsiness may occur with Dantrium therapy, and the concomitant administration of CNS depressants such as sedatives and tranquilizing agents may result in further drowsiness.
While a definite drug interaction with estrogen therapy has not yet been established, caution should be observed if the two drugs are to be given concomitantly. Hepatotoxicity has occurred more often in women over 35 years of age receiving concomitant estrogen therapy.
Cardiovascular collapse in patients treated simultaneously with verapamil and dantrolene sodium is rare. The combination of therapeutic doses of intravenous dantrolene sodium and verapamil in halothane/(alpha)-chloralose anesthetized swine has resulted in ventricular fibrillation and cardiovascular collapse in association with marked hyperkalemia. Until the relevance of these findings to humans is established, the combination of dantrolene sodium and calcium channel blockers is not recommended during the management of malignant hyperthermia.
Administration of Dantrium may potentiate vecuronium-induced neuromuscular block.
Dantrium should be used with caution in patients with impaired pulmonary function, particularly those with obstructive pulmonary disease, and in patients with severely impaired cardiac function due to myocardial disease. It should be used with caution in patients with a history of previous liver disease or dysfunction (see ).
Patients should be cautioned against driving a motor vehicle or participating in hazardous occupations while taking Dantrium . Caution should be exercised in the concomitant administration of tranquilizing agents.
Dantrium might possibly evoke a photosensitivity reaction; patients should be cautioned about exposure to sunlight while taking it.
The most frequently occurring side effects of Dantrium have been drowsiness, dizziness, weakness, general malaise, fatigue, and diarrhea. These are generally transient, occurring early in treatment, and can often be obviated by beginning with a low dose and increasing dosage gradually until an optimal regimen is established. Diarrhea may be severe and may necessitate temporary withdrawal of Dantrium therapy. If diarrhea recurs upon readministration of Dantrium , therapy should probably be withdrawn permanently.
Other less frequent side effects, listed according to system, are:
Gastrointestinal Constipation, rarely progressing to signs of intestinal obstruction, GI bleeding, anorexia, swallowing difficulty, gastric irritation, abdominal cramps, nausea and/or vomiting.
Hepatobiliary: Hepatitis (see ).
Neurologic: Speech disturbance, seizure, headache, light-headedness, visual disturbance, diplopia, alteration of taste, insomnia, drooling.
Cardiovascular Tachycardia, erratic blood pressure, phlebitis, heart failure.
Hematologic: Aplastic anemia, leukopenia, lymphocytic lymphoma, thrombocytopenia.
Psychiatric: Mental depression, mental confusion, increased nervousness.
Urogenital: Increased urinary frequency, crystalluria, hematuria, difficult erection, urinary incontinence and/or nocturia, difficult urination and/or urinary retention.
Integumentary: Abnormal hair growth, acne-like rash, pruritus, urticaria, eczematoid eruption, sweating.
Musculoskeletal: Myalgia, backache.
Respiratory: Feeling of suffocation, respiratory depression.
Special Senses: Excessive tearing.
Hypersensitivity: Pleural effusion with pericarditis, anaphylaxis.
Other: Chills and fever.
The published literature has included some reports of Dantrium use in patients with Neuroleptic Malignant Syndrome (NMS). Dantrium capsules are not indicated for the treatment of NMS and patients may expire despite treatment with Dantrium capsules.
Prior to the administration of Dantrium , consideration should be given to the potential response to treatment. A decrease in spasticity sufficient to allow a daily function not otherwise attainable should be the therapeutic goal of treatment with Dantrium . Refer to section for of response to be anticipated.
It is important to establish a therapeutic goal (regain and maintain a specific function such as therapeutic exercise program, utilization of braces, transfer maneuvers, etc.) before beginning Dantrium therapy. Dosage should be increased until the maximum performance compatible with the dysfunction due to underlying disease is achieved. No further increase in dosage is then indicated.
Usual Dosage: It is important that the dosage be titrated and individualized for maximum effect. The lowest dose compatible with optimal response is recommended.
In view of the potential for liver damage in long-term Dantrium use, therapy should be stopped if benefits are not evident within 45 days.
Adults: The following gradual titration schedule is suggested. Some patients will not respond until higher daily dosage is achieved. Each dosage level should be maintained for seven days to determine the patient' response. If no further benefit is observed at the next higher dose, dosage should be decreased to the previous lower dose.
25 mg once daily for seven days, then
25 mg t.i.d. for seven days
50 mg t.i.d. for seven days
100 mg t.i.d.
Therapy with a dose four times daily may be necessary for some individuals. Doses higher than 100 mg four times daily should not be used. (See Box Warning .)
Pediatric Patients: The following gradual titration schedule is suggested. Some patients will not respond until higher daily dosage is achieved. Each dosage level should be maintained for seven days to determine the patient' response. If no further benefit is observed at the next higher dose, dosage should be decreased to the previous lower dose.
0.5 mg/kg once daily for seven days, then
0.5 mg/kg t.i.d. for seven days
1 mg/kg t.i.d. for seven days
2 mg/kg t.i.d.
Therapy with a dose four times daily may be necessary for some individuals. Doses higher than 100 mg four times daily should not be used. (See Box Warning .)
Preoperatively: Administer 4 to 8 mg/kg/day of oral Dantrium in 3 or 4 divided doses for one or two days prior to surgery, with the last dose being given approximately 3 to 4 hours before scheduled surgery with a minimum of water.
This dosage will usually be associated with skeletal muscle weakness and sedation (sleepiness or drowsiness); adjustment can usually be made within the recommended dosage range to avoid incapacitation or excessive gastrointestinal irritation (including nausea and/or vomiting).
Oral Dantrium should also be administered following a malignant hyperthermia crisis, in doses of 4 to 8 mg/kg per day in four divided doses, for a one to three day period to prevent recurrence of the manifestations of malignant hyperthermia.
Symptoms which may occur in case of overdose include, but are not limited to, muscular weakness and alterations in the state of consciousness (e.g. lethargy, coma), vomiting, diarrhea, and crystalluria. For acute overdosage, general supportive measures should be employed along with immediate gastric lavage.
Intravenous fluids should be administered in fairly large quantities to avert the possibility of crystalluria. An adequate airway should be maintained and artificial resuscitation equipment should be at hand. Electrocardiographic monitoring should be instituted, and the patient carefully observed. To date, no experience has been reported with dialysis and its value in Dantrium overdosage is not known.
Dantrium (dantrolene sodium) is available in
25-mg opaque, orange and tan capsules:
NDC 0149-0030-05 bottle of 100
NDC 0149-0030-66 bottle of 500
NDC 0149-0030-77 hospital unit-dose strips in
boxes of 100
50-mg opaque, orange and tan capsules:
NDC 0149-0031-05 bottle of 100
100-mg opaque, orange and tan capsules:
NDC 0149-0033-05 bottle of 100
NDC 0149-0033-77 hospital unit-dose strips in
boxes of 100
Avoid excessive heat (over 104°F or 40°C).
Address medical inquiries to Procter & Gamble Pharmaceuticals, Medical Communications Department, 11450 Grooms Rd, Cincinnati, Ohio 45242.
CAUTION: Federal law prohibits dispensing without prescription.
Procter & Gamble Pharmaceuticals
Cincinnati, Ohio 45202
REVISED FEBRUARY 1997