 |
WARNING
|
* Data on file at Pharmacia & Upjohn
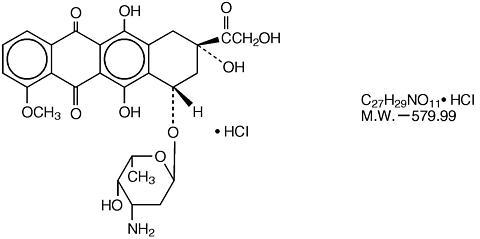
Doxorubicin is a cytotoxic anthracycline antibiotic isolated from cultures of Streptomyces peucetius var. caesius .
Doxorubicin consists of a naphthacenequinone nucleus linked through a glycosidic bond at ring atom 7 to an amino sugar, daunosamine.
Chemically, doxorubicin hydrochloride is:
5,12-Naphthacenedione, 10-[(3-amino-2,3,6-trideoxy-(alpha)-L- lyxo -hexopyranosyl)oxy]-7,8,9,10-tetrahydro- 6,8,11-trihydroxy-8-(hydroxylacetyl)-1-(methoxy-, hydrochloride (8 S-cis )-. The structural formula is as follows:
|
|
Doxorubicin binds to nucleic acids, presumably by specific intercalation of the planar anthracycline nucleus with the DNA double helix. The anthracycline ring is lipophilic, but the saturated end of the ring system contains abundant hydroxyl groups adjacent to the amino sugar, producing a hydrophilic center. The molecule is amphoteric, containing acidic functions in the ring phenolic groups and a basic function in the sugar amino group. It binds to cell membranes as well as plasma proteins.
ADRIAMYCIN RDF® (doxorubicin hydrochloride for injection, USP) a sterile red-orange lyophilized powder for intravenous use only, is available in 10, 20 and 50 mg single dose vials and a 150 mg multidose vial.
Each 10 mg single dose vial contains 10 mg of doxorubicin HCl, USP, 50 mg of lactose, NF (hydrous) and 1 mg of methylparaben, NF (added to enhance dissolution) as a sterile red-orange lyophilized powder.
Each 20 mg single dose vial contains 20 mg of doxorubicin HCl, USP, 100 mg of lactose, NF (hydrous) and 2 mg of methylparaben, NF (added to enhance dissolution) as a sterile red-orange lyophilized powder.
Each 50 mg single dose vial contains 50 mg of doxorubicin HCl, USP, 250 mg of lactose, NF (hydrous) and 5 mg of methylparaben, NF (added to enhance dissolution) as a sterile red-orange lyophilized powder.
Each 150 mg multidose vial contains 150 mg of doxorubicin HCl, USP, 750 mg of lactose, NF (hydrous) and 15 mg of methylparaben, NF (added to enhance dissolution) as a sterile red-orange lyophilized powder.
ADRIAMYCIN PFS® (doxorubicin hydrochloride injection, USP) is a sterile parenteral, isotonic solution for intravenous use only, containing no preservative, available in 5 mL (10 mg), 10 mL (20 mg), 25 mL (50 mg), and 37.5 mL (75 mg) single dose vials and a 100 mL (200 mg) multidose vial.
Each mL contains doxorubicin HCl 2 mg, USP and the following inactive ingredients: sodium chloride 0.9% and water for injection q.s. Hydrochloric acid is used to adjust the pH to a target pH of 3.0.
The cytotoxic effect of doxorubicin on malignant cells and its toxic effects on various organs are thought to be related to nucleotide base intercalation and cell membrane lipid binding activities of doxorubicin. Intercalation inhibits nucleotide replication and action of DNA and RNA polymerases. The interaction of doxorubicin with topoisomerase II to form DNA-cleavable complexes appears to be an important mechanism of doxorubicin cytocidal activity. Doxorubicin cellular membrane binding may effect a variety of cellular functions. Enzymatic electron reduction of doxorubicin by a variety of oxidases, reductases and dehydrogenases generate highly reactive species including the hydroxyl free radical OH·. Free radical formation has been implicated in doxorubicin cardiotoxicity by means of Cu (II) and Fe (III) reduction at the cellular level. Cells treated with doxorubicin have been shown to manifest the characteristic morphologic changes associated with apoptosis or programmed cell death. Doxorubicin-induced apoptosis may be an integral component of the cellular mechanism of action relating to therapeutic effects, toxicities, or both.
Animal studies have shown activity in a spectrum of experimental tumors, immunosuppression, carcinogenic properties in rodents, induction of a variety of toxic effects, including delayed and progressive cardiac toxicity, myelosuppression in all species and atrophy to testes in rats and dogs.
Pharmacokinetic studies, determined in patients with various types of tumors undergoing either single or multi-agent therapy have shown that doxorubicin follows a multiphasic disposition after intravenous injection. The initial distributive half-life of approximately 5.0 minutes suggests rapid tissue uptake of doxorubicin, while its slow elimination from tissues is reflected by a terminal half-life of 20 to 48 hours. Steady-state distribution volumes exceed 20 to 30 L/kg and are indicative of extensive drug uptake into tissues. Plasma clearance is in the range of 8 to 20 mL/min/kg and is predominately by metabolism and biliary excretion. Approximately 40% of the dose appears in the bile in 5 days, while only 5 to 12% of the drug and its metabolites appear in the urine during the same time period. Binding of doxorubicin and its major metabolite, doxorubicinol to plasma proteins is about 74 to 76% and is independent of plasma concentration of doxorubicin up to 2 µM. Enzymatic reduction at the 7 position and cleavage of the daunosamine sugar yields aglycones which are accompanied by free radical formation, the local production of which may contribute to the cardiotoxic activity of doxorubicin. Disposition of doxorubicinol (DOX-OL) in patients is formation rate limited. The terminal half-life of DOX-OL is similar to doxorubicin. The relative exposure of DOX-OL, compared to doxorubicin ranges between 0.4 to 0.6. In urine, <3% of the dose was recovered as DOX-OL over 7 days.
A published clinical study involving 6 men and 21 women with no prior anthracycline therapy reported a significantly higher median doxorubicin clearance in the men compared to the women (113 versus 45 L/hr). However, the terminal half-life of doxorubicin was longer in men compared to the women (54 versus 35 hrs.).
In four patients , dose-independent pharmacokinetics have been shown for doxorubicin in the dose range of 30 to 70 mg/m 2 . Systemic clearance of doxorubicin is significantly reduced in obese women with ideal body weight greater than 130%. There was a significant reduction in clearance without any change in volume of distribution in obese patients when compared with normal patients with less than 115% ideal body weight. The clearance of doxorubicin and doxorubicinol was also reduced in patients with impaired hepatic function. Doxorubicin was excreted in the milk of one lactating patient, with peak milk concentration at 24 hours after treatment being approximately 4.4-fold greater than the corresponding plasma concentration . Doxorubicin was detectable in the milk up to 72 hours after therapy with 70 mg/m 2 of doxorubicin given as a 15 minute intravenous infusion and 100 mg/m 2 of cisplatin as a 26 hour intravenous infusion. The peak concentration of doxorubicinol in milk at 24 hours was 0.2 µM and AUC up to 24 hours was 16.5 µM.hr while the AUC for doxorubicin was 9.9 µM.hr.
Following administration of 10 to 75-mg/m 2 doses of doxorubicin to 60 children and adolescents ranging from 2 months to 20 years of age, doxorubicin clearance average 1443 ± 114 mL/min/m 2 . Further analysis demonstrated that clearance in 52 children greater than 2 years of age (1540 mL/min/m 2 ) was increased compared with adults. However, clearance in infants younger than 2 years of age (813 mL/min/m 2 ) was decreased compared with older children and approached the range of clearance values determined in adults.
Doxorubicin does not cross the blood brain barrier.
ADRIAMYCIN PFS and ADRIAMYCIN RDF have been used successfully to produce regression in disseminated neoplastic conditions such as acute lymphoblastic leukemia, acute myeloblastic leukemia, Wilms' tumor, neuroblastoma, soft tissue and bone sarcomas, breast carcinoma, ovarian carcinoma, transitional cell bladder carinoma, thyroid carcinoma, gastric carcinoma, Hodgkin' disease, malignant lymphoma and bronchogenic carcinoma in which the small cell histologic type is the most responsive compared to other cell types.
Doxorubicin therapy should not be started in patients who have marked myelosuppression induced by previous treatment with other antitumor agents or by radiotherapy. Doxorubicin treatment is contraindicated in patients who received previous treatment with complete cumulative doses of doxorubicin, daunorubicin, idarubicin, and/or other anthracyclines and anthracenes.
Special attention must be given to the cardiotoxicity induced by doxorubicin. Irreversible myocardial toxicity, manifested in its most severe form by life-threatening or fatal congestive heart failure, may occur either during therapy or months to years after termination of therapy. The probability of developing impaired myocardial function, based on a combined index of signs, symptoms and decline in left ventricular ejection fraction (LVEF) is estimated to be 1 to 2% at a total cumulative dose of 300 mg/m 2 of doxorubicin, 3 to 5% at a dose of 400 mg/m 2 , 5 to 8% at a dose of 450 mg/m 2 and 6 to 20% at a dose of 500 mg/m 2 given in a schedule of a bolus injection once every 3 weeks (data on file at Pharmacia & Upjohn). In a retrospective review by Von Hoff et al, the probability of developing congestive heart failure was reported to be 5/168 (3%) at a cumulative dose of 430 mg/m 2 of doxorubicin, 8/110 (7%) at 575 mg/m 2 and 3/14 (21%) at 728 mg/m 2 . The cumulative incidence of CHF was 2.2%. In a prospective study of doxorubicin in combination with cyclophosphamide, fluorouracil and/or vincristine in patients with breast cancer or small cell lung cancer, the cumulative incidence of congestive heart failure was 5 to 6%. The probability of CHF at various cumulative doses of doxorubicin was 1.5% at 300 mg/m 2 , 4.9% at 400 mg/m 2 , 7.7% at 450 mg/m 2 and 20.5% at 500 mg/m 2 .
Cardiotoxicity may occur at lower doses in patients with prior mediastinal irradiation, concurrent cyclophosphamide therapy exposure at an early age and advanced age. Data also suggest that pre-existing heart disease is a co-factor for increased risk of doxorubicin cadiotoxicity. In such cases, cardiac toxicity may occur at doses lower than the respective recommended cumulative dose of doxorubicin. Studies have suggested that concomitant administration of doxorubicin and calcium channel entry blockers may increase the risk of doxorubicin cardiotoxicity. The total dose of doxorubicin administered to the individual patient should also take into account previous or concomitant therapy with related compounds such as daunorubicin, idarubicin and mitoxantrone. Cardiomyopathy and/or congestive heart failure may be encountered several months or years after discontinuation of doxorubicin therapy.
The risk of congestive heart failure and other acute manifestations of doxorubicin cardiotoxicity in pediatric patients may be as much or lower than in adults. Pediatric patients appear to be at particular risk for developing delayed cardiac toxicity in that doxorubicin induced cardiomyopathy impairs myocardial growth as pediatric patients mature, subsequently leading to possible development of congestive heart failure during early adulthood. As many as 40% of pediatric patients may have subclinical cardiac dysfunction and 5 to 10% of pediatric patients may develop congestive heart failure on long term follow-up. This late cardiac toxicity may be related to the dose of doxorubicin. The longer the length of follow-up the greater the increase in the detection rate.
Treatment of doxorubicin induced congestive heart failure includes the use of digitalis, diuretics, after load reducers such as angiotensin I converting enzyme (ACE) inhibitors, low salt diet, and bed rest. Such intervention may relieve symptoms and improve the functional status of the patient.
In adult patients severe cardiac toxicity may occur precipitously without antecedent ECG changes. Cardiomyopathy induced by anthracyclines is usually associated with very characteristic histopathologic changes on an endomyocardial biopsy (EM biopsy), and a decrease of left ventricular ejection fraction (LVEF), as measured by multi-gated radionuclide angiography (MUGA scans) and/or echocardiogram (ECHO), from pretreatment baseline values. However, it has not been demonstrated that monitoring of the ejection fraction will predict when individual patients are approaching their maximally tolerated cumulative dose of doxorubicin. Cardiac function should be carefully monitored during treatment to minimize the risk of cardiac toxicity. A baseline cardiac evaluation with an ECG, LVEF, and/or an echocardiogram (ECHO) is recommended especially in patients with risk factors for increased cardiac toxicity (pre-existing heart disease mediastinal irradiation, or concurrent cyclophosphamide therapy). Subsequent evaluations should be obtained at a cumulative dose of doxorubicin of at least 400 mg/m 2 and periodically thereafter during the course of therapy. Pediatric patients are at increased risk for developing delayed cardiotoxicity following doxorubicin administration and therefore at follow-up cardiac evaluation is recommended periodically to monitor for this delayed cardiotoxicity.
In adults, a 10% decline in LVEF to below the lower limit of normal or an absolute LVEF of 45%, or a 20% decline in LVEF at any level is indicative of deterioration in cardiac function. In pediatric patients, deterioration in cardiac function during or after the completion of therapy with doxorubicin is indicated by a drop in fractional shortening (FS) by an absolute value of >/=10 percentile units or below 29%, and a decline in LVEF of 10 percentile units or an LVEF below 55%. In general, if test results indicate deterioration in cardiac function associated with doxorubicin, the benefit of continued therapy should be carefully evaluated against the risk of producing irreversible cardiac damage.
Acute life-threatening arrhythmias have been reported to occur during or within a few hours after doxorubicin administration.
There is a high incidence of bone marrow depression, primarily of leukocytes, requiring careful hematologic monitoring. With the recommended dose schedule, leukopenia is usually transient, reaching its nadir 10 to 14 days after treatment with recovery usually occurring by the 21st day. White blood counts as low as 1000/mm 3 are to be expected during treatment with appropriate doses of doxorubicin. Red blood cell and platelet levels should also be monitored since they may also be depressed. Hematologic toxicity may require dose reduction or suspension or delay of doxorubicin therapy. Persistent severe myelosuppression may result in superinfection or hemorrhage.
Doxorubicin may potentiate the toxicity of other anticancer therapies. Exacerbation of cyclophosphamide induced hemorrhagic cystitis and enhancement of the hepatotoxicity of 6-mercaptopurine have been reported. Radiation induced toxicity to the myocardium, mucosae, skin and liver have been reported to be increased by the administration of doxorubicin. Pediatric patients receiving concomitant doxorubicin and actinomycin-D have manifested acute "recall" pneumonitis at variable times after local radiation therapy.
Since metabolism and excretion of doxorubicin occurs predominantly by the hepatobiliary route, toxicity to recommended doses of doxorubicin can be enhanced by hepatic impairment; therefore, prior to the individual dosing, evaluation of hepatic function is recommended using conventional laboratory tests such as SGOT, SGPT, alkaline phosphatase and bilirubin (see DOSAGE AND ADMINISTRATION ).
Necrotizing colitis manifested by typhlitis (cecal inflammation), bloody stools and severe and sometimes fatal infections have been associated with a combination of doxorubicin given by i.v. push daily for 3 days and cytarabine given by continuous infusion daily for 7 or more days.
On intravenous administration of doxorubicin, extravasation may occur with or without an accompanying stinging or burning sensation, even if blood returns well on aspiration of the infusion needle (see DOSAGE AND ADMINISTRATION ). If any signs or symptoms of extravasation have occurred, the injection or infusion should be immediately terminated and restarted in another vein.
Pregnancy Category D -- Safe use of doxorubicin in pregnancy has not been established. Doxorubicin is embryotoxic and teratogenic in rats and embryotoxic and abortifacient in rabbits. There are no adequate and well-controlled studies in pregnant women. If doxorubicin is to be used during pregnancy, or if the patient becomes pregnant during therapy, the patient should be apprised of the potential hazard to the fetus. Women of childbearing age should be advised to avoid becoming pregnant.
General
Doxorubicin is not an anti-microbial agent.
Information for Patients
ADRIAMYCIN PFS and ADRIAMYCIN RDF impart a red coloration to the urine for 1 to 2 days after administration, and patients should be advised to expect this during active therapy.
Paclitaxel Two published studies report that initial administration of paclitaxel infused over 24 hours followed by doxorubicin administered over 48 hours resulted in a significant decrease in doxorubicin clearance with more profound neutropenic and stomatitis episodes than the reverse sequence of administration.
Progesterone In a published study, progesterone was given intravenously to patients with advanced malignancies (ECOG PS<2) at high doses (up to 10 g over 24 hours) concomitantly with a fixed doxorubicin dose (60 mg/m 2 ) via bolus. Enhanced doxorubicin-induced neutropenia and thrombocytopenia were observed.
Verapamil A study of the effects of verapamil on the acute toxicity of doxorubicin in mice revealed higher initial peak concentrations of doxorubicin in the heart with a higher incidence and severity of degenerative changes in cardiac tissue resulting in a shorter survival.
Cyclosporine: The addition of cyclosporine to doxorubicin may result in increases in AUC for both doxorubicin and doxorubicinol possibly due to a decrease in clearance of parent drug and a decrease in metabolism of doxorubicinol. Literature reports suggest that adding cyclosporine to doxorubicin results in more profound and prolonged hematologic toxicity than doxorubicin alone. Coma and/or seizures have also been described.
Drug Interactions: Literature reports have also described the following drug interactions: phenobarbital increases the elimination of doxorubicin, phenytoin levels may be decreased by doxorubicin, streptozocin (Zanosar ® ) may inhibit hepatic metabolism of doxorubicin, and administration of live vaccines to immunosuppressed patients including those undergoing cytotoxic chemotherapy may be hazardous.
Laboratory Tests
Initial treatment with doxorubicin requires observation of the patient and periodic monitoring of complete blood counts, hepatic function tests, and radionuclide left ventricular ejection fraction. (See ).
Like other cytotoxic drugs, doxorubicin may induce "tumor lysis syndrome" and hyperuricemia in patients with rapidly growing tumors. Appropriate supportive and pharmacologic measures may prevent or alleviate this complication.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Formal long-term carcinogenicity studies have not been conducted with doxorubicin. Doxorubicin and related compounds have been shown to have mutagenic and carcinogenic properties when tested in experimental models (including bacterial systems, mammalian cells in culture, and female Sprague-Dawley rats).
The possible adverse effect on fertility in males and females in humans or experimental animals have not been adequately evaluated. Testicular atrophy was observed in rats and dogs.
A variant of chemotherapy-related acute non-lymphocytic leukemia has been reported to occur infrequently a few years after multiple drug treatment of some neoplasms, which sometimes included doxorubicin. The exact role of doxorubicin has not been elucidated. Pediatric patients treated with doxorubicin or other topoisomerase II inhibitors are at a risk for developing acute myelogenous leukemia and other neoplasms. The extent of increased risk associated with doxorubicin has not been precisely quantified.
Pregnancy Category D
Nursing Mothers:
Because of the potential for serious adverse reactions in nursing infants from doxorubicin, mothers should be advised to discontinue nursing during doxorubicin therapy.
Pediatric Use:
Pediatric patients are at increased risk for developing delayed cardiotoxicity. Follow-up cardiac evaluations are recommended periodically to monitor for this delayed cardiotoxicity (see ).
Doxorubicin, as a component of intensive chemotherapy regimens administered to pediatric patients, may contribute to prepubertal growth failure. It may also contribute to gonadal impairment, which is usually temporary.
Dose limiting toxicities of therapy are myelosuppression and cardiotoxicity. Other reactions reported are:
Cutaneous--Reversible complete alopecia occurs in most cases. Hyperpigmentation of nailbeds and dermal crease, primarily in pediatric patients, and onycholysis have been reported in a few cases. Recall of skin reaction due to prior radiotherapy has occurred with doxorubicin administration.
Gastrointestinal--Acute nausea and vomiting occurs frequently and may be severe. This may be alleviated by antiemetic therapy. Mucositis (stomatitis and esophagitis) may occur 5 to 10 days after administration. The effect may be severe leading to ulceration and represents a site of origin for severe infections. The dosage regimen consisting of administration of doxorubicin on three successive days results in greater incidence and severity of mucositis. Ulceration and necrosis of the colon, especially the cecum, may occur leading to bleeding or severe infections which can be fatal. This reaction has been reported in patients with acute nonlymphocytic leukemia treated with a 3-day course of doxorubicin combined with cytarabine. Anorexia and diarrhea have been occasionally reported.
Vascular--Phlebosclerosis has been reported especially when small veins are used or a single vein is used for repeated administration. Facial flushing may occur if the injection is given too rapidly.
Local--Severe cellulitis, vesication and tissue necrosis will occur if extravasation of doxorubicin occurs during administration. Erythematous streaking along the vein proximal to the site of injection had been reported (see DOSAGE AND ADMINISTRATION ).
Hematologic--The occurrence of secondary acute myeloid leukemia with or without a preleukemic phase has been reported rarely in patients concurrently treated with doxorubicin in association with DNA-damaging antineoplastic agents. Such cases could have a short (1-3 years) latency period. Pediatric patients are also at risk of developing secondary acute myeloid leukemia.
Hypersensitivity--Fever, chills and urticaria have been reported occasionally. Anaphylaxis may occur. A case of apparent cross sensitivity to lincomycin has been reported.
Neurological--Peripheral neurotoxicity in the form of local-regional sensory and/or motor disturbances have been reported in patients treated intra-arterially with doxorubicin, mostly in combination with cisplatin. Animal studies have demonstrated seizures and coma in rodents and dogs treated with intra-carotid doxorubicin. Seizures and coma have been reported in patients treated with doxorubicin in combination with cisplatin or vincristine.
Other--Conjunctivitis and lacrimation occur rarely.
Acute overdosage with doxorubicin enhances the toxic effect of mucositis, leukopenia and thrombocytopenia. Treatment of acute overdosage consists of treatment of the severely myelosuppressed patient with hospitalization, antimicrobials, platelet transfusions and symptomatic treatment of mucositis. Use of hemopoietic growth factor (G-CSF, GM-CSF) may be considered.
The 150 mg ADRIAMYCIN RDF and the 100 mL (2 mg/mL) ADRIAMYCIN PFS vials are packaged as multiple dose vials and caution should be exercised to prevent inadvertent overdosage.
Cumulative dosage with doxorubicin increases the risk of cardiomyopathy and resultant congestive heart failure (see ). Treatment consists of vigorous management of congestive heart failure with digitalis preparations, diuretics, and after-load reducers such as ACE inhibitors.
Care in the administration of ADRIAMYCIN PFS and ADRIAMYCIN RDF will reduce the chance of perivenous infiltration (see ). It may also decrease the change of local reactions such as urticaria and erythematous streaking. On intravenous administration of doxorubicin, extravasation may occur with or without an accompanying burning or stinging sensation, even if blood returns well on aspiration of the infusion needle. If any signs or symptoms of extravasation have occurred, the injection or infusion should be immediately terminated and restarted in another vein. If extravasation is suspected, intermittent application of ice to the site for 15 min. q.i.d. × 3 days may be useful. The benefit of local administration of drugs has not been clearly established. Because of the progressive nature of extravasation reactions, close observation and plastic surgery consultation is recommended. Blistering, ulceration and/or persistent pain are indications for wide excision surgery, followed by split-thickness skin grafting.
The most commonly used dose schedule when used as a single agent is 60 to 75 mg/m 2 as a single intravenous injection administered at 21-day intervals. The lower dosage should be given to patients with inadequate marrow reserves due to old age, or prior therapy, or neoplastic marrow infiltration. ADRIAMYCIN PFS and ADRIAMYCIN RDF have been used concurrently with other approved chemotherapeutic agents. Evidence is available that in some types of neoplastic disease combination chemotherapy is superior to single agents. The benefits and risks of such therapy continue to be elucidated. When used in combination with other chemotherapy drugs, the most commonly used dosage of doxorubicin is 40 to 60 mg/m 2 given as a single intravenous injection every 21 to 28 days. Doxorubicin dosage must be reduced in case of hyperbilirubinemia as follows:
|
Reconstitution Directions: ADRIAMYCIN RDF 10 mg, 20 mg, 50 mg, and 150 mg vials should be reconstituted with 5 mL, 10 mL, 25 mL, and 75 mL, respectively, of Sodium Chloride Injection, USP (0.9%), to give a final concentration of 2 mg/mL of doxorubicin hydrochloride. An appropriate volume of air should be withdrawn from the vial during reconstitution to avoid excessive pressure buildup. Bacteriostatic diluents are not recommended.
After adding the diluent, the vial should be shaken and the contents allowed to dissolve. The reconstituted solution is stable for 7 days at room temperature and under normal room light (100 foot-candles) and 15 days under refrigeration (2° to 8°C). It should be protected from exposure to sunlight. Discard any of the unused solution from the 10 mg, 20 mg, and 50 mg single dose vials. Unused solutions of the multiple dose vial remaining beyond the recommended storage times should be discarded.
It is recommended that ADRIAMYCIN PFS and ADRIAMYCIN RDF be slowly administered into the tubing of a freely running intravenous infusion of Sodium Chloride Injection, USP, or 5% Dextrose Injection, USP. The tubing should be attached to a Butterfly® needle inserted preferably into a large vein. If possible, avoid veins over joints or in extremities with compromised venous or lymphatic drainage. The rate of administration is dependent on the size of the vein, and the dosage. However, the dose should be administered in not less than 3 to 5 minutes. Local erythematous streaking along the vein as well as facial flushing may be indicated of too rapid an administration. A burning or stinging sensation may be indicative of perivenous infiltration and the infusion should be immediately terminated and restarted in another vein. Perivenous infiltration may occur painlessly.
Doxorubicin should not be mixed with heparin or fluorouracil since it has been reported that these drugs are incompatible to the extent that a precipitate may form. Until specific compatibility data are available, it is not recommended that doxorubicin be mixed with other drugs.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
Handling and Disposal: Skin reactions associated with doxorubicin have been reported. Skin accidentally exposed to doxorubicin should be rinsed copiously with soap and warm water, and if the eyes are involved, standard irrigation techniques should be used immediately. The use of goggles, gloves, and protective gowns is recommended during preparation and administration of the drug.
Procedures for proper handling and disposal of anti-cancer drugs should be considered. Several guidelines on this subject have been published. 1-7 There is no general agreement that all the procedures recommended in the guidelines are necessary or appropriate.
Caregivers of pediatric patients receiving doxorubicin should be counseled to take precautions (such as wearing latex gloves) to prevent contact with the patient' urine and other body fluids for at least 5 days after each treatment.
ADRIAMYCIN RDF® Powder for Injection (doxorubicin hydrochloride for injection, USP) is available as follows:
NDC 0013-1086-91 10 mg single dose vial, 10 vial packs
NDC 0013-1096-91 20 mg single dose vial, 10 vial packs
NDC 0013-1106-79 50 mg single dose vial, single packs
Store at controlled room temperature, 15° to 30°C (59° to 86°F). Protect from light.
Retain in carton until time of use. Contains no preservative. Discard unused portion.
MULTIDOSE VIAL:
NDC 0013-1116-83 150 mg multidose vial, single packs
Store at controlled room temperature, 15° to 30°C (59° to 86°F). Protect from light.
Retain in carton until time of use.
After adding the diluent, the vial should be shaken and the contents allowed to dissolve. The reconstituted solution is stable for 7 days at room temperature and under normal room light (100 foot-candles) and 15 days under refrigeration (2° to 8°C). It should be protected from exposure to sunlight. Discard any unused solution from the 10 mg, 20 mg and 50 mg single dose vials. Unused solutions of the multiple dose vial remaining beyond the recommended storage times should be discarded.
Pharmacia & Upjohn S.p.A.
Milan, Italy
ADRIAMYCIN PFS® Injection (doxorubicin hydrochloride injection, USP)
SINGLE DOSE VIALS:
Sterile single use only, contains no preservative.
NDC 0013-1136-91 10 mg vial, 2 mg/mL, 5 mL, 10 vial packs
NDC 0013-1146-91 20 mg vial, 2 mg/mL, 10 mL, 10 vial packs
NDC 0013-1156-79 50 mg vial, 2 mg/mL, 25 mL, single vial packs
NDC 0013-1176-87 75 mg vial, 2 mg/mL, 37.5 mL, single vial packs
Store refrigeration, 2° to 8°C (36° to 46°F). Protect from light. Retain in carton until time of use. Discard unused portion.
MULTIDOSE VIAL:
Sterile multidose vial, contains no preservative.
NDC 0013-1166-83 200 mg, 2 mg/mL, 100 mL, multidose vial, single vial packs
Store refrigeration, 2° to 8°C (36° to 46°F). Protect from light. Retain in carton until contents are used.
Pharmacia & Upjohn S.p.A.
Milan, Italy
SP Pharmaceuticals LLC
Albuquerque, NM 87109, USA
Rx only
Pharmacia & Upjohn Company, Kalamazoo, MI 49001, USA
817 336 203 Revised April 2000