 |
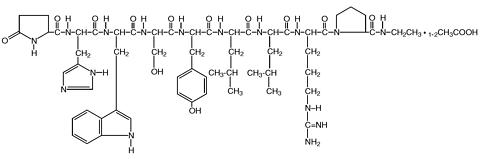
Leuprolide acetate is a synthetic nonapeptide analog of naturally occurring gonadotropin-releasing hormone (GnRH or LH-RH). The analog possesses greater potency than the natural hormone. The chemical name is 5-oxo-L-prolyl-L-histidyl-L-tryptophyl-L-seryl-L-tyrosyl-D-leucyl-L-leucyl-L-arginyl-N-ethyl-L-prolinamide acetate (salt) with the following structural formula:
|
|
LUPRON DEPOT-3 Month 22.5 mg is available in a prefilled dual-chamber syringe containing sterile lyophilized microspheres which, when mixed with diluent, become a suspension intended as an intramuscular injection to be given ONCE EVERY THREE MONTHS (84 days).
The front chamber of LUPRON DEPOT-3 Month 22.5 mg prefilled dual-chamber syringe contains leuprolide acetate (22.5 mg), polylactic acid (198.6 mg) and D-mannitol (38.9 mg). The second chamber of diluent contains carboxymethylcellulose sodium (7.5 mg), D-mannitol (75.0 mg), polysorbate 80 (1.5 mg), water for injection, USP, and glacial acetic acid, USP to control pH.
During the manufacture of LUPRON DEPOT-3 Month 22.5 mg, acetic acid is lost, leaving the peptide.
Leuprolide acetate, an LH-RH agonist, acts as a potent inhibitor of gonadotropin secretion when given continuously and in therapeutic doses. Animal and human studies indicate that following an initial stimulation, chronic administration of leuprolide acetate results in suppression of ovarian and testicular steroidogenesis. This effect is reversible upon discontinuation of drug therapy. Administration of leuprolide acetate has resulted in inhibition of the growth of certain hormone dependent tumors (prostatic tumors in Noble and Dunning male rats and DMBA-induced mammary tumors in female rats) as well as atrophy of the reproductive organs.
In humans, administration of leuprolide acetate results in an initial increase in circulating levels of luteinizing hormone (LH) and follicle stimulating hormone (FSH), leading to a transient increase in levels of the gonadal steroids (testosterone and dihydrotestosterone in males, and estrone and estradiol in premenopausal females). However, continuous administration of leuprolide acetate results in decreased levels of LH and FSH. In males, testosterone is reduced to castrate levels. In premenopausal females, estrogens are reduced to postmenopausal levels. These decreases occur within two to four weeks after initiation of treatment, and castrate levels of testosterone in prostatic cancer patients have been demonstrated for more than five years. Leuprolide acetate is not active when given orally.
Absorption Following a single injection of the three month formulation of LUPRON DEPOT-3 Month 22.5 mg in patients, mean peak plasma leuprolide concentration of 48.9 ng/mL was observed at 4 hours and then declined to 0.67 ng/mL at 12 weeks. Leuprolide appeared to be released at a constant rate following the onset of steady-state levels during the third week after dosing, providing steady plasma concentrations through the 12-week dosing interval. However, intact leuprolide and an inactive major metabolite could not be distinguished by the assay which was employed in the study. Detectable levels of leuprolide were present at all measurement points in all patients. The initial burst, followed by the rapid decline to a steady-state level, was similar to the release pattern seen with the monthly formulation.
Distribution The mean steady-state volume of distribution of leuprolide following intravenous bolus administration to healthy male volunteers was 27 L. In vitro binding to human plasma proteins ranged from 43% to 49%.
Metabolism In healthy male volunteers, a 1 mg bolus of leuprolide administered intravenously revealed that the mean systemic clearance was 7.6 L/h, with a terminal elimination half-life of approximately 3 hours based on a two compartment model.
In rats and dogs, administration of 14 C-labeled leuprolide was shown to be metabolized to smaller inactive peptides, a pentapeptide (Metabolite I), tripeptides (Metabolites II and III) and a dipeptide (Metabolite IV). These fragments may be further catabolized.
The major metabolite (M-I) plasma concentrations measured in 5 prostate cancer patients reached maximum concentration 2 to 6 hours after dosing and were approximately 6% of the peak parent drug concentration. One week after dosing, mean plasma M-I concentrations were approximately 20% of mean leuprolide concentrations.
Excretion Following administration of LUPRON DEPOT 3.75 mg to 3 patients, less than 5% of the dose was recovered as parent and M-I metabolite in the urine.
Special Populations The pharmacokinetics of the drug in hepatically and renally impaired patients have not been determined.
In clinical studies, serum testosterone was suppressed to castrate within 30 days in 87 of 92 (95%) patients and within an additional two weeks in three patients. Two patients did not suppress for 15 and 28 weeks, respectively. Suppression was maintained in all of these patients with the exception of transient minimal testosterone elevations in one of them, and in another an increase in serum testosterone to above the castrate range was recorded during the 12 hour observation period after a subsequent injection. This represents stimulation of gonadotropin secretion.
An 85% rate of "no progression" was achieved during the initial 24 weeks of treatment. A decrease from baseline in serum PSA of >/=90% was reported in 71% of the patients and a change to within the normal range (</=3.99 ng/mL) in 63% of the patients.
Periodic monitoring of serum testosterone and PSA levels is recommended, especially if the anticipated clinical or biochemical response to treatment has not been achieved. It should be noted that results of testosterone determinations are dependent on assay methodology. It is advisable to be aware of the type and precision of the assay methodology to make appropriate clinical and therapeutic decisions.
LUPRON DEPOT-3 Month 22.5 mg is indicated in the palliative treatment of advanced prostatic cancer. It offers an alternative treatment of prostatic cancer when orchiectomy or estrogen administration are either not indicated or unacceptable to the patient. In clinical trials, the safety and efficacy of LUPRON DEPOT-3 Month 22.5 mg were similar to that of the original daily subcutaneous injection and the monthly depot formulation.
A report of an anaphylactic reaction to synthetic GnRH (Factrel) has been reported in the medical literature. 1
LUPRON DEPOT is contraindicated in women who are or may become pregnant while receiving the drug. When administered on day 6 of pregnancy at test dosages of 0.00024, 0.0024, and 0.024 mg/kg (1/600 to 1/6 of the human dose) to rabbits, the monthly formulation of LUPRON DEPOT produced a dose-related increase in major fetal abnormalities. Similar studies in rats failed to demonstrate an increase in fetal malformations. There was increased fetal mortality and decreased fetal weights with the two higher doses of the monthly formulation of LUPRON DEPOT in rabbits and with the highest dose in rats. The effects on fetal mortality are logical consequences of the alterations in hormonal levels brought about by this drug. Therefore, the possibility exists that spontaneous abortion may occur if the drug is administered during pregnancy.
Isolated cases of worsening of signs and symptoms during the first weeks of treatment have been reported with LH-RH analogs. Worsening of symptoms may contribute to paralysis with or without fatal complications. For patients at risk, the physician may consider initiating therapy with daily LUPRON ® (leuprolide acetate) Injection for the first two weeks to facilitate withdrawal of treatment if that is considered necessary.
General Patients with metastatic vertebral lesions and/or with urinary tract obstruction should be closely observed during the first few weeks of therapy. (See section.)
Laboratory Tests Response to LUPRON DEPOT-3 Month 22.5 mg should be monitored by measuring serum levels of testosterone, as well as prostate-specific antigen and prostatic acid phosphatase. In the majority of patients, testosterone levels increased above baseline during the first week, declining thereafter to baseline levels or below by the end of the second week. Castrate levels were reached within two to four weeks and once achieved were maintained for as long as the patients received their injections.
Drug Interactions No pharmacokinetic-based drug-drug interaction studies have been conducted with LUPRON DEPOT. However, because leuprolide acetate is a peptide that is primarily degraded by peptidase and not by cytochrome P-450 enzymes as noted in specific studies, and the drug is only about 46% bound to plasma proteins, drug interactions would not be expected to occur.
Drug/Laboratory Test Interactions Administration of LUPRON DEPOT 3.75 mg in women results in suppression of the pituitary-gonadal system. Normal function is usually restored within one to three months after treatment is discontinued. Therefore, diagnostic tests of pituitary gonadotropic and gonadal functions conducted during treatment and up to three months after discontinuation of LUPRON DEPOT 3.75 mg therapy may be misleading.
Carcinogenesis, Mutagenesis, Impairment of Fertility Two-year carcinogenicity studies were conducted in rats and mice. In rats, a dose-related increase of benign pituitary hyperplasia and benign pituitary adenomas was noted at 24 months when the drug was administered subcutaneously at high daily doses (0.6 to 4 mg/kg). There was a significant but not dose-related increase of pancreatic islet-cell adenomas in females and of testicular interstitial cell adenomas in males (highest incidence in the low dose group). In mice no pituitary abnormalities were observed at a dose as high as 60 mg/kg for two years. Patients have been treated with leuprolide acetate for up to three years with doses as high as 10 mg/day and for two years with doses as high as 20 mg/day without demonstrable pituitary abnormalities.
Mutagenicity studies have been performed with leuprolide acetate using bacterial and mammalian systems. These studies provided no evidence of a mutagenic potential.
Clinical and pharmacologic studies in adults (>/=18 years) with leuprolide acetate and similar analogs have shown reversibility of fertility suppression when the drug is discontinued after continuous administration for periods of up to 24 weeks.
Pregnancy, Teratogenic Effects
Pregnancy Category X. (See CONTRAINDICATIONS section.)
Pediatric Use See LUPRON DEPOT-PED® (leuprolide acetate for depot suspension) labeling for the safety and effectiveness of the monthly formulation in children with central precocious puberty.
In the majority of patients testosterone levels increased above baseline during the first week, declining thereafter to baseline levels or below by the end of the second week of treatment.
Potential exacerbations of signs and symptoms during the first few weeks of treatment is a concern in patients with vertebral metastases and/or urinary obstruction or hematuria which, if aggravated, may lead to neurological problems such as temporary weakness and/or paresthesia of the lower limbs or worsening of urinary symptoms. (See section.)
In two clinical trials of LUPRON DEPOT-3 Month 22.5 mg, the following adverse reactions were reported to have a possible or probable relationship to drug as ascribed by the treating physician in 5% or more of the patients receiving the drug. Often, causality is difficult to assess in patients with metastatic prostate cancer. Reactions considered not drug-related are excluded.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In these same studies, the following adverse reactions were reported in less than 5% of the patients on LUPRON DEPOT-3 Month 22.5 mg.
Body As A Whole -- Enlarged abdomen, Fever; Cardiovascular System-- Arrhythmia, Bradycardia, Heart failure, Hypertension, Hypotension, Varicose vein; Digestive System --Anorexia, Duodenal ulcer, Increased appetite, Thirst/dry mouth; Hemic and Lymphatic System --Anemia, Lymphedema; Metabolic and Nutritional Disorders --Dehydration, Edema; Central/Peripheral Nervous System --Anxiety, Delusions, Depression, Hypesthesia, Libido decreased*, Nervousness, Paresthesia; Respiratory System --Epistaxis, Pharyngitis, Pleural effusion, Pneumonia; Special Senses --Abnormal vision, Amblyopia, Dry eyes, Tinnitus; Urogenital System --Gynecomastia, Impotence*, Penis disorders, Testis disorders.
Laboratory Abnormalities of certain parameters were observed, but are difficult to assess in this population. The following were recorded in >/= 5% of patients: Increased BUN, Hyperglycemia, Hyperlipidemia (total cholesterol, LDL-cholesterol, triglycerides), Hyperphosphatemia, Abnormal liver function tests, Increased PT, Increased PTT. Additional laboratory abnormalities reported were: Decreased platelets, Decreased potassium and Increased WBC.
*Physiologic effect of decreased testosterone.
Postmarketing
During postmarketing surveillance, which includes other dosage forms, the following adverse events were reported.
Symptoms consistent with an anaphylactoid or asthmatic process have been rarely reported. Rash, urticaria, and photosensitivity reactions have also been reported.
Localized reactions including induration and abscess have been reported at the site of injection.
Hemic and Lymphatic System --Decreased WBC; Central/Peripheral Nervous System --Peripheral neuropathy, Spinal fracture/paralysis; Musculoskeletal System --Tenosynovitis-like symptoms; Urogenital System -- Prostate pain.
See other LUPRON DEPOT and LUPRON Injection package inserts for other events reported in different patient populations.
In rats subcutaneous administration of 250 to 500 times the recommended human dose, expressed on a per body weight basis, resulted in dyspnea, decreased activity, and local irritation at the injection site. There is no evidence at present that there is a clinical counterpart of this phenomenon. In early clinical trials with daily subcutaneous leuprolide acetate, doses as high as 20 mg/day for up to two years caused no adverse effects differing from those observed with the 1 mg/day dose.
LUPRON DEPOT Must Be Administered Under The Supervision Of A Physician.
The recommended dose of LUPRON DEPOT-3 Month 22.5 mg to be administered is one injection every three months (84 days). Due to different release characteristics, a fractional dose of this 3-month depot formulation is not equivalent to the same dose of the monthly formulation and should not be given.
Incorporated in a depot formulation, the lyophilized microspheres are to be reconstituted and administered every three months as a single intramuscular injection, in accord with the following directions:
Although the potency of the reconstituted suspension has been shown to be stable for 24 hours, since the product does not contain a preservative, the suspension should be discarded if not used immediately.
As with other drugs administered by injection, the injection site should be varied periodically.
LUPRON DEPOT-3 Month 22.5 mg is packaged as follows:
Kit with prefilled
dual-chamber syringe (NDC 0300-3346-01)
Each syringe contains sterile lyophilized microspheres which is leuprolide acetate incorporated in a biodegradable polymer of polylactic acid. When mixed with 1.5 mL of accompanying diluent, LUPRON DEPOT-3 Month 22.5 mg is administered as a single IM injection EVERY THREE MONTHS (84 days).
An information pamphlet for patients is included with the kit.
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [see USP Controlled Room Temperature]
Rx only
U.S. Patent Nos. 4,652,441; 4,728,721; 4,849,228; 4,917,893; 4,954,298; 5,330,767; 5,476,663; 5,480,656; 5,575,987; 5,631,020; 5,631,021; 5,643,607; and 5,716,640.
Manufactured for
TAP Pharmaceuticals Inc.
Lake Forest, IL 60045, U.S.A.
by Takeda Chemical Industries, Ltd. Osaka, JAPAN 541
®--Registered trademark
(No. 3346)
03-5028-R5; Revised: March 2000
© 1995-2000, TAP Pharmaceutical Products Inc.
 |