 |
WARNINGCO-ADMINISTRATION OF NORVIR WITH CERTAIN NONSEDATING ANTIHISTAMINES, SEDATIVE HYPNOTICS, ANTIARRHYTHMICS, OR ERGOT ALKALOID PREPARATIONS MAY RESULT IN POTENTIALLY SERIOUS AND/OR LIFE-THREATENING ADVERSE EVENTS DUE TO POSSIBLE EFFECTS OF NORVIR ON THE HEPATIC METABOLISM OF CERTAIN DRUGS. SEE CONTRAINDICATIONS AND PRECAUTIONS SECTIONS. |
NORVIR (ritonavir) is an inhibitor of HIV protease with activity against the Human Immunodeficiency Virus (HIV).
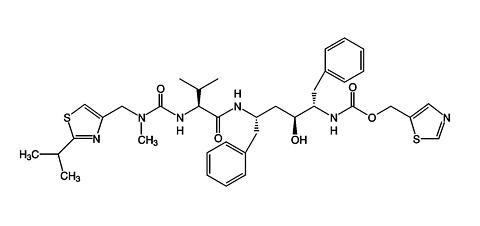
Ritonavir is chemically designated as 10-Hydroxy-2-methyl-5-(1-methylethyl)-1-[2-(1-methylethyl)-4-thiazolyl]-3, 6-dioxo-8,11-bis(phenylmethyl)-2,4,7,12-tetraazatridecan-13-oic acid, 5-thiazolylmethyl ester, [5S-(5R*,8R*,10R*,11R*)]. Its molecular formula is C 37 H 48 N 6 O 5 S 2 , and its molecular weight is 720.95. Ritonavir has the following structural formula:
|
|
Ritonavir is a white-to-light-tan powder. Ritonavir has a bitter metallic taste. It is freely soluble in methanol and ethanol, soluble in isopropanol and practically insoluble in water.
NORVIR soft gelatin capsules are available for oral administration in a strength of 100 mg ritonavir with the following inactive ingredients: Butylated hydroxytoluene, ethanol, gelatin, iron oxide, oleic acid, polyoxyl 35 castor oil, and titanium dioxide.
NORVIR oral solution is available for oral administration as 80 mg/mL of ritonavir in a peppermint and caramel flavored vehicle. Each 8-ounce bottle contains 19.2 grams of ritonavir. NORVIR oral solution also contains ethanol, water, polyoxyl 35 castor oil, propylene glycol, anhydrous citric acid to adjust pH, saccharin sodium, peppermint oil, creamy caramel flavoring, and FD&C Yellow No. 6.
Mechanism of action : Ritonavir is a peptidomimetic inhibitor of both the HIV-1 and HIV-2 proteases. Inhibition of HIV protease renders the enzyme incapable of processing the gag-pol polyprotein precursor which leads to production of non-infectious immature HIV particles.
Antiviral activity in vitro : The activity of ritonavir was assessed in vitro in acutely infected lymphoblastoid cell lines and in peripheral blood lymphocytes. The concentration of drug that inhibits 50% (EC 50 ) of viral replication ranged from 3.8 to 153 nM depending upon the HIV-1 isolate and the cells employed. The average EC 50 for low passage clinical isolates was 22 nM (n=13). In MT 4 cells, ritonavir demonstrated additive effects against HIV-1 in combination with either zidovudine (ZDV) or didanosine (ddI). Studies which measured cytotoxicity of ritonavir on several cell lines showed that >20 µM was required to inhibit cellular growth by 50% resulting in an in vitro therapeutic index of at least 1000.
Resistance : HIV-1 isolates with reduced susceptibility to ritonavir have been selected in vitro . Genotypic analysis of these isolates showed mutations in the HIV protease gene at amino acid positions 84 (Ile to Val), 82 (Val to Phe), 71 (Ala to Val), and 46 (Met to Ile). Phenotypic (n=18) and genotypic (n=44) changes in HIV isolates from selected patients treated with ritonavir were monitored in phase I/II trials over a period of 3 to 32 weeks. Mutations associated with the HIV viral protease in isolates obtained from 41 patients appeared to occur in a stepwise and ordered fashion; in sequence, these mutations were position 82 (Val to Ala/Phe), 54 (Ile to Val), 71 (Ala to Val/Thr), and 36 (Ile to Leu), followed by combinations of mutations at an additional 5 specific amino acid positions. Of 18 patients for which both phenotypic and genotypic analysis were performed on free virus isolated from plasma, 12 showed reduced susceptibility to ritonavir in vitro . All 18 patients possessed one or more mutations in the viral protease gene. The 82 mutation appeared to be necessary but not sufficient to confer phenotypic resistance. Phenotypic resistance was defined as a >/=5-fold decrease in viral sensitivity in vitro from baseline. The clinical relevance of phenotypic and genotypic changes associated with ritonavir therapy has not been established.
Cross-resistance to other antiretrovirals : Among protease inhibitors variable cross-resistance has been recognized. Serial HIV isolates obtained from six patients during ritonavir therapy showed a decrease in ritonavir susceptibility in vitro but did not demonstrate a concordant decrease in susceptibility to saquinavir in vitro when compared to matched baseline isolates. However, isolates from two of these patients demonstrated decreased susceptibility to indinavir in vitro (8-fold). Isolates from 5 patients were also tested for cross-resistance to amprenavir and nelfinavir; isolates from 2 patients had a decrease in susceptibility to nelfinavir (12- to 14-fold), and none to amprenavir. Cross-resistance between ritonavir and reverse transcriptase inhibitors is unlikely because of the different enzyme targets involved. One ZDV-resistant HIV isolate tested in vitro retained full susceptibility to ritonavir.
The pharmacokinetics of ritonavir have been studied in healthy volunteers and HIV-infected patients (CD 4 >/= 50 cells/µL). See Table 1 for ritonavir pharmacokinetic characteristics.
The absolute bioavailability of ritonavir has not been determined. After a 600 mg dose of oral solution, peak concentrations of ritonavir were achieved approximately 2 hours and 4 hours after dosing under fasting and non-fasting (514 KCal; 9% fat, 12% protein, and 79% carbohydrate) conditions, respectively. When the oral solution was given under non-fasting conditions, peak ritonavir concentrations decreased 23% and the extent of absorption decreased 7% relative to fasting conditions. Dilution of the oral solution, within one hour of administration, with 240 mL of chocolate milk, Advera® or Ensure® did not significantly affect the extent and rate of ritonavir absorption. After a single 600 mg dose under non-fasting conditions, in two separate studies, the soft gelatin capsule (n=57) and oral solution (n=18) formulations yielded mean ± SD areas under the plasma concentration-time curve (AUCs) of 121.7 ± 53.8 and 129.0 ± 39.3 µg·h/mL, respectively. Relative to fasting conditions, the extent of absorption of ritonavir from the soft gelatin capsule formulation was 13% higher when administered with a meal (615 KCal; 14.5% fat, 9% protein, and 76% carbohydrate).
Nearly all of the plasma radioactivity after a single oral 600 mg dose of 14 C-ritonavir oral solution (n=5) was attributed to unchanged ritonavir. Five ritonavir metabolites have been identified in human urine and feces. The isopropylthiazole oxidation metabolite (M-2) is the major metabolite and has antiviral activity similar to that of parent drug; however, the concentrations of this metabolite in plasma are low. In vitro studies utilizing human liver microsomes have demonstrated that cytochrome P450 3A (CYP3A) is the major isoform involved in ritonavir metabolism, although CYP2D6 also contributes to the formation of M-2.
In a study of five subjects receiving a 600 mg dose of 14 C-ritonavir oral solution, 11.3 ± 2.8% of the dose was excreted into the urine, with 3.5 ± 1.8% of the dose excreted as unchanged parent drug. In that study, 86.4 ± 2.9% of the dose was excreted in the feces with 33.8 ± 10.8% of the dose excreted as unchanged parent drug. Upon multiple dosing, ritonavir accumulation is less than predicted from a single dose possibly due to a time and dose-related increase in clearance.
|
|||||||||||||||||||||||||||||||||||||||
The pharmacokinetic profile of ritonavir in pediatric patients below the age of 2 years has not been established. Steady-state pharmacokinetics were evaluated in 37 HIV-infected patients ages 2 to 14 years receiving doses ranging from 250 mg/m 2 b.i.d. to 400 mg/m 2 b.i.d. Across dose groups, ritonavir steady-state oral clearance (CL/F/m 2 ) was approximately 1.5 times faster in pediatric patients than in adult subjects. Ritonavir concentrations obtained after 350 to 400 mg/m 2 twice daily in pediatric patients were comparable to those obtained in adults receiving 600 mg (approximately 330 mg/m 2 ) twice daily.
Gender, Race and Age : No age-related pharmacokinetic differences have been observed in adult patients (18 to 63 years). Ritonavir pharmacokinetics have not been studied in older patients. A study of ritonavir pharmacokinetics in healthy males and females showed no statistically significant differences in the pharmacokinetics of ritonavir. Pharmacokinetic differences due to race have not been identified.
Renal Insufficiency : Ritonavir pharmacokinetics have not been studied in patients with renal insufficiency; however, since renal clearance is negligible, a decrease in total body clearance is not expected in patients with renal insufficiency.
Hepatic Insufficiency : Ritonavir pharmacokinetics have not been studied in subjects with hepatic insufficiency (see PRECAUTIONS ).
Drug-Drug Interactions : Table 2 summarizes the effects on AUC and C max , with 95% confidence intervals (95% CI), of co-administration of ritonavir with a variety of drugs. For information about clinical recommendations see PRECAUTIONS -- Drug Interactions .
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
NORVIR is indicated in combination with other antiretroviral agents for the treatment of HIV-infection. This indication is based on the results from a study in patients with advanced HIV disease that showed a reduction in both mortality and AIDS-defining clinical events for patients who received NORVIR either alone or in combination with nucleoside analogues. Median duration of follow-up in this study was 13.5 months.
The activity of NORVIR as monotherapy or in combination with nucleoside analogues has been evaluated in 1446 patients enrolled in two double-blind, randomized trials.
Study 247 was a randomized, double-blind trial (with open-label follow-up) conducted in HIV-infected patients with at least nine months of prior antiretroviral therapy and baseline CD 4 cell counts </= 100 cells/µL. NORVIR 600 mg b.i.d. or placebo was added to each patient' baseline antiretroviral therapy regimen, which could have consisted of up to two approved antiretroviral agents. The study accrued 1090 patients, with mean baseline CD 4 cell count at study entry of 32 cells/µL. After the clinical benefit of NORVIR therapy was demonstrated, all patients were eligible to switch to open-label NORVIR for the duration of the follow-up period. Median duration of double-blind therapy with NORVIR and placebo was 6 months. The median duration of follow-up through the end of the open-label phase was 13.5 months for patients randomized to NORVIR and 14 months for patients randomized to placebo.
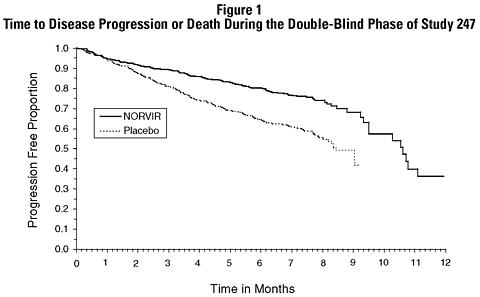
The cumulative incidence of clinical disease progression or death during the double-blind phase of Study 247 was 26% for patients initially randomized to NORVIR compared to 42% for patients initially randomized to placebo. This difference in rates was statistically significant (see Figure 1).
 |
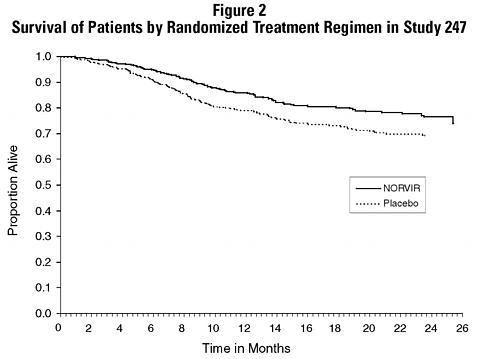
The cumulative mortality through the end of the open-label follow-up phase for patients enrolled in Study 247 was 18% for patients initially randomized to NORVIR compared to 26% for patients initially randomized to placebo. This difference in rates was statistically significant (see Figure 2). Since the analysis at the end of the open-label phase includes patients in the placebo arm who were switched from placebo to NORVIR therapy, the survival benefit of NORVIR cannot be precisely estimated.
 |
Figures 3 and 4 summarize the mean change from baseline for CD 4 cell count and plasma HIV RNA (copies/mL), respectively, during the first 24 weeks for the double-blind phase of Study 247.
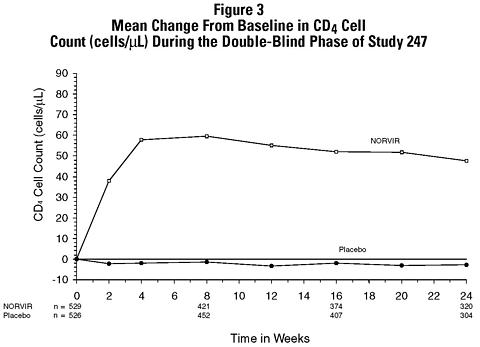 |
 |
In Study 245, 356 antiretroviral-naive HIV-infected patients (mean baseline CD 4 = 364 cells/µL) were randomized to receive either NORVIR 600 mg b.i.d., zidovudine 200 mg t.i.d., or a combination of these drugs. Figures 5 and 6 summarize the mean change from baseline for CD 4 cell count and plasma HIV RNA (copies/mL), respectively, during the first 24 weeks for the double-blind phase of Study 245.
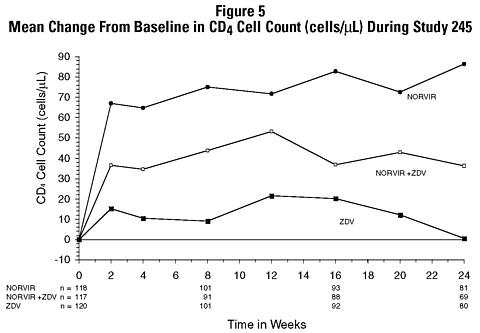 |
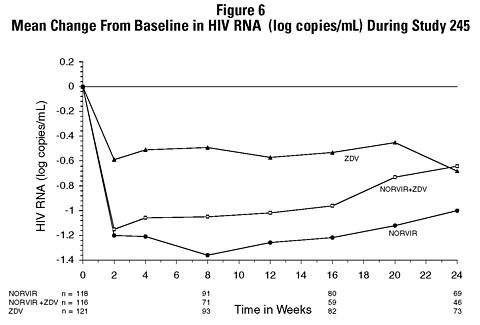 |
NORVIR is contraindicated in patients with known hypersensitivity to ritonavir or any of its ingredients.
NORVIR should not be administered concurrently with the drugs listed in Table 3 (also see PRECAUTIONS Table 4: Contraindicated Drugs) because competition for primarily CYP3A by ritonavir could result in inhibition of the metabolism of these drugs and create the potential for serious and/or life-threatening reactions such as cardiac arrhythmias, prolonged or increased sedation, and respiratory depression.
Postmarketing reports indicate that co-administration of ritonavir with ergotamine or dihydroergotamine has been associated with acute ergot toxicity characterized by peripheral vasospasm and ischemia of the extremities.
|
The magnitude of the interactions and therapeutic consequences between ritonavir and the drugs listed in Table 4 Predicted Drug Interactions: Use With Caution cannot be predicted with any certainty. When co-administering ritonavir with any agent listed in Table 4 Predicted Drug Interactions: Use With Caution , special attention is warranted.
Cardiac and neurologic events have been reported with ritonavir when co-administered with disopyramide, mexiletine, nefazodone, fluoxetine and beta blockers. The possibility of drug interaction cannot be excluded.
Particular caution should be used when prescribing sildenafil in patients receiving NORVIR. Co-administration of NORVIR with sildenafil is expected to substantially increase sildenafil concentrations (11-fold increase in AUC) and may result in an increase in sildenafil-associated adverse events, including hypotension, syncope, visual changes, and prolonged erection (see PRECAUTIONS : Drug Interactions , Table 4 Established Drug Interactions: Alteration in Dose or Regimen Recommended Based on Drug Interaction Studies and the complete prescribing information for sildenafil).
Concomitant use of NORVIR with lovastatin or simvastatin is not recommended. Caution should be exercised if HIV protease inhibitors, including NORVIR, are used concurrently with other HMG-CoA reductase inhibitors that are also metabolized by the CYP3A4 pathway (e.g., atorvastatin or cerivastatin). The risk of myopathy including rhabdomyolysis may be increased when HIV protease inhibitors, including NORVIR, are used in combination with these drugs.
Concomitant use of NORVIR, and St. John's wort (hypericum perforatum) or products containing St. John's wort is not recommended. Coadministration of protease inhibitors, including NORVIR, with St. John's wort is expected to substantially decrease protease inhibitor concentrations and may result in sub-optimal levels of NORVIR and lead to loss of virologic response and possible resistance to NORVIR or to the class of protease inhibitors.
Allergic reactions including urticaria, mild skin eruptions, bronchospasm, and angioedema have been reported. Rare cases of anaphylaxis and Stevens-Johnson syndrome have also been reported.
Hepatic transaminase elevations exceeding 5 times the upper limit of normal, clinical hepatitis, and jaundice have occurred in patients receiving NORVIR alone or in combination with other antiretroviral drugs (see Table 6). There may be an increased risk for transaminase elevations in patients with underlying hepatitis B or C. Therefore, caution should be exercised when administering NORVIR to patients with pre-existing liver diseases, liver enzyme abnormalities, or hepatitis. Increased AST/ALT monitoring should be considered in these patients, especially during the first three months of NORVIR treatment.
There have been postmarketing reports of hepatic dysfunction, including some fatalities. These have generally occurred in patients taking multiple concomitant medications and/or with advanced AIDS.
Pancreatitis has been observed in patients receiving NORVIR therapy, including those who developed hypertriglyceridemia. In some cases fatalities have been observed. Patients with advanced HIV disease may be at increased risk of elevated triglycerides and pancreatitis.
Pancreatitis should be considered if clinical symptoms (nausea, vomiting, abdominal pain) or abnormalities in laboratory values (such as increased serum lipase or amylase values) suggestive of pancreatitis should occur. Patients who exhibit these signs or symptoms should be evaluated and NORVIR therapy should be discontinued if a diagnosis of pancreatitis is made.
New onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus, and hyperglycemia have been reported during postmarketing surveillance in HIV-infected patients receiving protease inhibitor therapy. Some patients required either initiation or dose adjustments of insulin or oral hypoglycemic agents for treatment of these events. In some cases, diabetic ketoacidosis has occurred. In those patients who discontinued protease inhibitor therapy, hyperglycemia persisted in some cases. Because these events have been reported voluntarily during clinical practice, estimates of frequency cannot be made and a causal relationship between protease inhibitor therapy and these events has not been established.
Ritonavir is principally metabolized by the liver. Therefore, caution should be exercised when administering this drug to patients with impaired hepatic function (see ).
Varying degrees of cross-resistance among protease inhibitors have been observed. Continued administration of ritonavir therapy following loss of viral suppression may increase the likelihood of cross-resistance to other protease inhibitors (see MICROBIOLOGY ).
There have been reports of increased bleeding, including spontaneous skin hematomas and hemarthrosis, in patients with hemophilia type A and B treated with protease inhibitors. In some patients additional factor VIII was given. In more than half of the reported cases, treatment with protease inhibitors was continued or reintroduced. A causal relationship has not been established.
Redistribution/accumulation of body fat including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, breast enlargement, and "cushingoid appearance" have been observed in patients receiving protease inhibitors. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
Treatment with NORVIR therapy alone or in combination with saquinavir has resulted in substantial increases in the concentration of total triglycerides and cholesterol. Triglyceride and cholesterol testing should be performed prior to initiating NORVIR therapy and at periodic intervals during therapy. Lipid disorders should be managed as clinically appropriate. See PRECAUTIONS Table 4 for additional information on potential drug interactions with NORVIR and HMG CoA reductase inhibitors.
Patients should be informed that NORVIR is not a cure for HIV infection and that they may continue to acquire illnesses associated with advanced HIV infection, including opportunistic infections.
Patients should be told that the long-term effects of NORVIR are unknown at this time. They should be informed that NORVIR therapy has not been shown to reduce the risk of transmitting HIV to others through sexual contact or blood contamination.
Patients should be advised to take NORVIR with food, if possible.
Patients should be informed to take NORVIR every day as prescribed. Patients should not alter the dose or discontinue NORVIR without consulting their doctor. If a dose is missed, patients should take the next dose as soon as possible. However, if a dose is skipped, the patient should not double the next dose.
Patients should be informed that redistribution or accumulation of body fat may occur in patients receiving protease inhibitors and that the cause and long-term health effects of these conditions are not known at this time.
NORVIR may interact with some drugs; therefore, patients should be advised to report to their doctor the use of any other prescription, non-prescription medication or herbal products, particularly St. John's wort.
Ritonavir has been shown to increase triglycerides, cholesterol, SGOT (AST), SGPT (ALT), GGT, CPK, and uric acid. Appropriate laboratory testing should be performed prior to initiating NORVIR therapy and at periodic intervals or if any clinical signs or symptoms occur during therapy. For comprehensive information concerning laboratory test alterations associated with nucleoside analogues, physicians should refer to the complete product information for each of these drugs.
Ritonavir has been found to be an inhibitor of cytochrome P450 3A (CYP3A) both in vitro and in vivo (Table 2). Agents that are extensively metabolized by CYP3A and have high first pass metabolism appear to be the most susceptible to large increases in AUC (>3-fold) when co-administered with ritonavir. Ritonavir also inhibits CYP2D6 to a lesser extent. Co-administration of substrates of CYP2D6 with ritonavir could result in increases (up to 2-fold) in the AUC of the other agent, possibly requiring a proportional dosage reduction. Ritonavir also appears to induce CYP3A as well as other enzymes, including glucuronosyl transferase, CYP1A2, and possibly CYP2C9.
Drugs that are contraindicated specifically due to the expected magnitude of interaction and potential for serious adverse events are listed both in CONTRAINDICATIONS Table 3 and under Contraindicated Drugs in Table 4.
Those drug interactions that have been established based on drug interaction studies are listed with the pharmacokinetic results in CLINICAL PHARMACOLOGY , Table 2. The clinical recommendations based on the results of these studies are listed in Table 4 Established Drug Interactions: Alteration in Dose or Regimen Recommended Based on Drug Interaction Studies.
A systematic review of over 200 medications prescribed to HIV-infected patients was performed to identify potential drug interactions with ritonavir. 2 There are a number of agents in which CYP3A or CYP2D6 partially contribute to the metabolism of the agent. In these cases, the magnitude of the interaction and therapeutic consequences cannot be predicted with any certainty.
When co-administering ritonavir with calcium channel blockers, immunosuppressants, some HMG-CoA reductase inhibitors (see , Drug Interactions ), some steroids, or other substrates of CYP3A, or most antidepressants, certain antiarrhythmics, and some narcotic analgesics which are partially mediated by CYP2D6 metabolism, it is possible that substantial increases in concentrations of these other agents may occur, possibly requiring a dosage reduction (>50%); examples are listed in Table 4 Predicted Drug Interactions: Use With Caution, Dose Decrease May be Needed.
When co-administering ritonavir with any agent having a narrow therapeutic margin, such as anticoagulants, anticonvulsants, and antiarrhythmics, special attention is warranted. With some agents, the metabolism may be induced, resulting in decreased concentrations (see Table 4 Predicted Drug Interactions: Use With Caution, Dose Increase May be Needed).
|
||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||
Cardiac and neurologic events have been reported when ritonavir has been co-administered with disopyramide, mexiletine, nefazodone, fluoxetine, and beta blockers. The possibility of drug interaction cannot be excluded.
Long-term carcinogenicity studies of ritonavir in animal systems have not been completed. However, ritonavir was not mutagenic or clastogenic in a battery of in vitro and in vivo assays including bacterial reverse mutation (Ames) using S. typhimurium and E. coli , mouse lymphoma, mouse micronucleus, and chromosome aberrations in human lymphocytes.
Pregnancy Category B: Ritonavir produced no effects on fertility in rats at drug exposures approximately 40% (male) and 60% (female) of that achieved with the proposed therapeutic dose. Higher dosages were not feasible due to hepatic toxicity.
No treatment-related malformations were observed when ritonavir was administered to pregnant rats or rabbits. Developmental toxicity observed in rats (early resorptions, decreased fetal body weight and ossification delays and developmental variations) occurred at a maternally toxic dosage at an exposure equivalent to approximately 30% of that achieved with the proposed therapeutic dose. A slight increase in the incidence of cryptorchidism was also noted in rats at an exposure approximately 22% of that achieved with the proposed therapeutic dose.
Developmental toxicity observed in rabbits (resorptions, decreased litter size and decreased fetal weights) also occurred at a maternally toxic dosage equivalent to 1.8 times the proposed therapeutic dose based on a body surface area conversion factor.
There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers: It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when ritonavir is administered to a nursing woman. However, the U.S. Public Health Service Centers for Disease Control and Prevention advises HIV-infected women not to breast-feed to avoid postnatal transmission of HIV to a child who may not be infected.
The safety and pharmacokinetic profile of ritonavir in pediatric patients below the age of 2 years have not been established. In HIV-infected patients age 2 to 16 years, the adverse event profile seen during a clinical trial and postmarketing experience was similar to that for adult patients. The evaluation of the antiviral activity of ritonavir in pediatric patients in clinical trials is ongoing.
The safety of NORVIR alone and in combination with nucleoside analogues was studied in 1270 patients. Table 5 lists treatment-emergent adverse events (at least possibly related and of at least moderate intensity) that occurred in 2% or greater of patients receiving NORVIR alone or in combination with nucleosides in Study 245 or Study 247 and in combination with saquinavir in ongoing Study 462. In that study, 141 protease inhibitor-naive, HIV-infected patients with mean baseline CD 4 of 300 cells/µL were randomized to one of four regimens of NORVIR + saquinavir, including NORVIR 400 mg b.i.d. + saquinavir 400 mg b.i.d. Overall the most frequently reported clinical adverse events, other than asthenia, among patients receiving NORVIR were gastrointestinal and neurological disturbances including nausea, diarrhea, vomiting, anorexia, abdominal pain, taste perversion, and circumoral and peripheral paresthesias. Similar adverse event profiles were reported in patients receiving ritonavir in other trials.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Adverse events occurring in less than 2% of patients receiving NORVIR in all phase II/phase III studies and considered at least possibly related or of unknown relationship to treatment and of at least moderate intensity are listed below by body system.
Body as a Whole: Abdomen enlarged, accidental injury, allergic reaction, back pain, cachexia, chest pain, chills, facial edema, facial pain, flu syndrome, hormone level altered, hypothermia, kidney pain, neck pain, neck rigidity, pelvic pain, photosensitivity reaction, and substernal chest pain.
Cardiovascular System: Cardiovascular disorder, cerebral ischemia, cerebral venous thrombosis, hypertension, hypotension, migraine, myocardial infarct, palpitation, peripheral vascular disorder, phlebitis, postural hypotension, tachycardia and vasospasm.
Digestive System: Abnormal stools, bloody diarrhea, cheilitis, cholestatic jaundice, colitis, dry mouth, dysphagia, eructation, esophageal ulcer, esophagitis, gastritis, gastroenteritis, gastrointestinal disorder, gastrointestinal hemorrhage, gingivitis, hepatic coma, hepatitis, hepatomegaly, hepatosplenomegaly, ileus, liver damage, melena, mouth ulcer, pancreatitis, pseudomembranous colitis, rectal disorder, rectal hemorrhage, sialadenitis, stomatitis, tenesmus, thirst, tongue edema, and ulcerative colitis.
Endocrine System: Adrenal cortex insufficiency and diabetes mellitus.
Hemic and Lymphatic System: Acute myeloblastic leukemia, anemia, ecchymosis, leukopenia, lymphadenopathy, lymphocytosis, myeloproliferative disorder, and thrombocytopenia.
Metabolic and Nutritional Disorders: Albuminuria, alcohol intolerance, avitaminosis, BUN increased, dehydration, edema, enzymatic abnormality, glycosuria, gout, hypercholesteremia, peripheral edema, and xanthomatosis.
Musculoskeletal System: Arthritis, arthrosis, bone disorder, bone pain, extraocular palsy, joint disorder, leg cramps, muscle cramps, muscle weakness, myositis, and twitching.
Nervous System: Abnormal dreams, abnormal gait, agitation, amnesia, aphasia, ataxia, coma, convulsion, dementia, depersonalization, diplopia, emotional lability, euphoria, grand mal convulsion, hallucinations, hyperesthesia, hyperkinesia, hypesthesia, incoordination, libido decreased, manic reaction, nervousness, neuralgia, neuropathy, paralysis, peripheral neuropathic pain, peripheral neuropathy, peripheral sensory neuropathy, personality disorder, sleep disorder, speech disorder, stupor, subdural hematoma, tremor, urinary retention, vertigo, and vestibular disorder.
Respiratory System: Asthma, bronchitis, dyspnea, epistaxis, hiccup, hypoventilation, increased cough, interstitial pneumonia, larynx edema, lung disorder, rhinitis, and sinusitis.
Skin and Appendages: Acne, contact dermatitis, dry skin, eczema, erythema multiforme, exfoliative dermatitis, folliculitis, fungal dermatitis, furunculosis, maculopapular rash, molluscum contagiosum, onychomycosis, pruritus, psoriasis, pustular rash, seborrhea, skin discoloration, skin disorder, skin hypertrophy, skin melanoma, urticaria, and vesiculobullous rash.
Special Senses: Abnormal electro-oculogram, abnormal electroretinogram, abnormal vision, amblyopia/blurred vision, blepharitis, conjunctivitis, ear pain, eye disorder, eye pain, hearing impairment, increased cerumen, iritis, parosmia, photophobia, taste loss, tinnitus, uveitis, visual field defect, and vitreous disorder.
Urogenital System: Acute kidney failure, breast pain, cystitis, dysuria, hematuria, impotence, kidney calculus, kidney failure, kidney function abnormal, kidney pain, menorrhagia, penis disorder, polyuria, urethritis, urinary frequency, urinary tract infection, and vaginitis.
Post-Marketing Experience:
There have been postmarketing reports of seizure. Cause and effect relationship has not been established.
Dehydration, usually associated with gastrointestinal symptoms, and sometimes resulting in hypotension, syncope, or renal insufficiency has been reported. Syncope, orthostatic hypotension, and renal insufficiency have also been reported without known dehydration.
Redistribution/accumulation of body fat has been reported (see PRECAUTIONS , Fat Redistribution ). There have been reports of increased bleeding in patients with hemophilia A or B (see PRECAUTIONS , Hemophilia ).
Table 6 shows the percentage of patients who developed marked laboratory abnormalities.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Human Overdose Experience: Human experience of acute overdose with NORVIR is limited. One patient in clinical trials took NORVIR 1500 mg/day for two days. The patient reported paresthesias which resolved after the dose was decreased. A post-marketing case of renal failure with eosinophilia has been reported with ritonavir overdose.
The approximate lethal dose was found to be greater than 20 times the related human dose in rats and 10 times the related human dose in mice.
Treatment of overdose with NORVIR consists of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient. There is no specific antidote for overdose with NORVIR. If indicated, elimination of unabsorbed drug should be achieved by emesis or gastric lavage; usual precautions should be observed to maintain the airway. Administration of activated charcoal may also be used to aid in removal of unabsorbed drug. Since ritonavir is extensively metabolized by the liver and is highly protein bound, dialysis is unlikely to be beneficial in significant removal of the drug. A Certified Poison Control Center should be consulted for up-to-date information on the management of overdose with NORVIR.
NORVIR is administered orally. It is recommended that NORVIR be taken with meals if possible. Patients may improve the taste of NORVIR oral solution by mixing with chocolate milk, Ensure®, or Advera® within one hour of dosing. The effects of antacids on the absorption of ritonavir have not been studied.
The recommended dosage of ritonavir is 600 mg twice daily by mouth. Use of a dose titration schedule may help to reduce treatment-emergent adverse events while maintaining appropriate ritonavir plasma levels. Ritonavir should be started at no less than 300 mg twice daily and increased at 2 to 3 day intervals by 100 mg twice daily. If saquinavir and ritonavir are used in combination, the dosage of saquinavir should be reduced to 400 mg twice daily. The optimum dosage of NORVIR (400 mg or 600 mg twice daily), in combination with saquinavir, has not been determined; however, the combination regimen was better tolerated in patients who received NORVIR 400 mg twice daily.
Ritonavir should be used in combination with other antiretroviral agents (see General Dosing Guidelines ). The recommended dosage of ritonavir is 400 mg/m 2 twice daily by mouth and should not exceed 600 mg twice daily. Ritonavir should be started at 250 mg/m 2 and increased at 2 to 3 day intervals by 50 mg/m 2 twice daily. If patients do not tolerate 400 mg/m 2 twice daily due to adverse events, the highest tolerated dose may be used for maintenance therapy in combination with other antiretroviral agents; however, alternative therapy should be considered. When possible, dose should be administered using a calibrated dosing syringe.
|
|||||||||||||||||||||||||||||||||||||||||||||
Patients should be aware that frequently observed adverse events, such as mild to moderate gastrointestinal disturbances and paraesthesias, may diminish as therapy is continued. In addition, patients initiating combination regimens with NORVIR and nucleosides may improve gastrointestinal tolerance by initiating NORVIR alone and subsequently adding nucleosides before completing two weeks of NORVIR monotherapy.
NORVIR (ritonavir capsules) soft gelatin are white capsules imprinted with the corporate logo
![]() ,100 and the Abbo-Code DS, available in the following package size:
,100 and the Abbo-Code DS, available in the following package size:
Bottles of 120 capsules each ..................... ( NDC 0074-6633-22).
Recommended storage: Store soft gelatin capsules in the refrigerator between 36-46°F (2-8°C) until dispensed. Refrigeration of NORVIR soft gelatin capsules by the patient is recommended, but not required if used within 30 days and stored below 77°F (25°C). Protect from light. Avoid exposure to excessive heat.
NORVIR (ritonavir oral solution) is an orange-colored liquid, supplied in amber-colored, multi-dose bottles containing 600 mg ritonavir per 7.5 mL marked dosage cup (80 mg/mL) in the following size:
240 mL bottles ..........................................( NDC 0074-1940-63).
Recommended storage: Store NORVIR oral solution at room temperature 68°F to 77°F (20°C to 25°C). Do not refrigerate. Shake well before each use. Use by product expiration date.
Product should be stored and dispensed in the original container.
Avoid exposure to excessive heat. Keep cap tightly closed.
NORVIR oral solution is manufactured by Abbott Laboratories, North Chicago, IL 60064, U.S.A. or Abbott Laboratories LTD, Queenborough, Kent, England. Distributed by Abbott Laboratories, North Chicago, IL 60064, U.S.A.
 |
 |
Revised: March, 2000
ABBOTT LABORATORIES
NORTH CHICAGO, IL 60064, U.S.A.
Please read this leaflet carefully before you start taking NORVIR. Also, read it each time you get your NORVIR prescription refilled, just in case something has changed. Remember that this information does not take the place of careful discussions with your doctor when you start this medication and at check ups.
You should remain under a doctor' care when taking NORVIR and you should not change or stop treatment without first talking with your doctor.
You should tell your doctor about any drug you are taking or planning to take because taking NORVIR with some medications can result in serious or life-threatening problems.
Talk to your doctor if you have any questions about NORVIR. Your doctor or pharmacist can also give you more information about NORVIR.
NORVIR is in a class of drugs called the HIV protease (PRO-tee-ase) inhibitors. NORVIR is used in combination with other anti-HIV drugs to treat people with human immunodeficiency virus (HIV) infection. HIV infection leads to the destruction of CD 4 (T) cells, which are important to the immune system. After a large number of CD 4 (T) cells have been destroyed, acquired immune deficiency syndrome (AIDS) develops.
NORVIR works by blocking HIV protease (a protein-cutting enzyme), which is required for HIV to multiply. NORVIR has been shown to significantly reduce the amount of HIV in the blood and increase the number of CD 4 (T) cells. Patients who took NORVIR in clinical studies had significant reductions in both death and AIDS defining diseases; however NORVIR may not have these effects in all patients.
NORVIR is not a cure for HIV infection or AIDS. The long-term effects of NORVIR are not known at this time. People taking NORVIR may still develop opportunistic infections or other conditions associated with HIV infection. Some of these conditions are pneumonia, herpes virus infections, and Mycobacterium avium complex (MAC) infections.
NORVIR does not reduce the risk of passing HIV to others through sexual contact or blood contamination. Continue to practice safe sex and do not use or share dirty needles.
It is important that you do not miss any doses. If you miss a dose of NORVIR, take it as soon as possible and then take your next scheduled dose at its regularly scheduled time. If it is almost time for your next dose, wait and take the next dose at the regularly scheduled time. Do not double the next dose.
Together with your doctor, you need to decide whether NORVIR is appropriate for you.
NORVIR may interact with other drugs, including those you take without a prescription. You must tell your doctor about all the drugs you are taking or are planning to take before you take NORVIR.
Cordarone® (amiodarone)
Ergotamine and dihydroergotamine such as Cafergot®, Migranal®, D.H.E 45®, and others
Halcion® (triazolam)
Hismanal® (astemizole)
Orap® (pimozide)
Propulsid® (cisapride)
Quinidine, also known as Quinaglute®, Cardioquin®, Quinidex®, and others
Rythmol® (propafenone)
Seldane® (terfenadine)
Tambocor® (flecainide)
Vascor® (bepridil)
Versed® (midazolam)
It is possible that your doctor may need to increase or decrease the dose of other drugs when you are also taking NORVIR. Remember to tell your doctor all drugs you are taking or plan to take.
Mycobutin® (rifabutin) Your doctor will lower your dose of Mycobutin
Viagra® (sildenafil)
Before you take Viagra with NORVIR, talk to your doctor about possible drug interactions and side effects. If you take Viagra and NORVIR together, you may be at risk of side effects of Viagra such as low blood pressure, visual changes, and penile erection lasting more than 4 hours. If an erection lasts longer than 4 hours, you should get medical help immediately to avoid permanent damage to your penis. Your doctor can explain these symptoms to you.
The following drug reduces blood levels of NORVIR:
Rifampin, also known as Rimactane®, Rifadin®, Rifater®, or Rifamate®
There have been other side effects noted in patients receiving NORVIR; however, these side effects may have been due to other drugs that patients were taking or to the illness itself. If you have questions about side effects, ask your doctor, nurse, or pharmacist. You should report any new or persistent symptoms to your doctor immediately.
Do not keep medicine that is out of date or that you no longer need. Be sure that if you throw any medicine away, it is out of the reach of children.
If you would like more information about NORVIR, ask your doctor or pharmacist. If you have any questions or concerns about taking NORVIR, talk with your doctor.
If you suspect that you took more than the prescribed dose of this medicine, contact your local poison control center or emergency room immediately.
Ref. 03-5026-R14
Revised: March, 2000
ABBOTT LABORATORIES
NORTH CHICAGO, IL 60064, U.S.A.