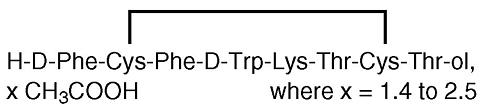 |
The following prescribing information is based on official labeling in effect May 1999.
Sandostatin® (octreotide acetate) Injection, a cyclic octapeptide prepared as a clear sterile solution of octreotide, acetate salt, in a buffered lactic acid solution for administration by deep subcutaneous (intrafat) or intravenous injection. Octreotide acetate, known chemically as L-Cysteinamide, D-phenylalanyl-L-cysteinyl-L-phenylalanyl-D-tryptophyl-L-lysyl-L-threonyl-N-[2-hydroxy-1-(hydroxymethyl)propyl]-, cyclic (2->7)-disulfide; [R-(R*, R*)] acetate salt, is a long-acting octapeptide with pharmacologic actions mimicking those of the natural hormone somatostatin.
Sandostatin® (octreotide acetate) Injection is available as: sterile 1 mL ampuls in 3 strengths, containing 50, 100, or 500 mcg octreotide (as acetate), and sterile 5 mL multi-dose vials in 2 strengths, containing 200 and 1000 mcg/mL of octreotide (as acetate).
Each ampul also contains:
lactic acid, USP.................................................. 3.4 mg
mannitol, USP..................................................... 45 mg
sodium bicarbonate, USP.................. qs to pH 4.2 ± 0.3
water for injection, USP................................ qs to 1 mL
Each mL of the multi-dose vials also contains:
lactic acid, USP.................................................. 3.4 mg
mannitol, USP...................................................... 45 mg
phenol, USP....................................................... 5.0 mg
sodium bicarbonate, USP.................. qs to pH 4.2 ± 0.3
water for injection, USP.................................qs to 1 mL
Lactic acid and sodium bicarbonate are added to provide a buffered solution, pH to 4.2 ± 0.3.
The molecular weight of octreotide acetate is 1019.3 (free peptide, C 49 H 66 N 10 O 10 S 2 ) and its amino acid sequence is:
|
|
Sandostatin® (octreotide acetate) exerts pharmacologic actions similar to the natural hormone, somatostatin. It is an even more potent inhibitor of growth hormone, glucagon, and insulin than somatostatin. Like somatostatin, it also suppresses LH response to GnRH, decreases splanchnic blood flow, and inhibits release of serotonin, gastrin, vasoactive intestinal peptide, secretin, motilin, and pancreatic polypeptide.
By virtue of these pharmacological actions, Sandostatin® (octreotide acetate) has been used to treat the symptoms associated with metastatic carcinoid tumors (flushing and diarrhea), and Vasoactive Intestinal Peptide (VIP) secreting adenomas (watery diarrhea).
Sandostatin® (octreotide acetate) substantially reduces growth hormone and/or IGF-I (somatomedin C) levels in patients with acromegaly.
Single doses of Sandostatin® (octreotide acetate) have been shown to inhibit gallbladder contractility and to decrease bile secretion in normal volunteers. In controlled clinical trials the incidence of gallstone or biliary sludge formation was markedly increased (See ).
Sandostatin® (octreotide acetate) suppresses secretion of thyroid stimulating hormone (TSH).
After subcutaneous injection, octreotide is absorbed rapidly and completely from the injection site. Peak concentrations of 5.2 ng/mL (100 mcg dose) were reached 0.4 hours after dosing. Using a specific radioimmunoassay, intravenous and subcutaneous doses were found to be bioequivalent. Peak concentrations and area under the curve values were dose proportional both after subcutaneous or intravenous single doses up to 400 mcg and with multiple doses of 200 mcg t.i.d. (600 mcg/day). Clearance was reduced by about 66% suggesting non-linear kinetics of the drug at daily doses of 600 mcg/day as compared to 150 mcg/day. The relative decrease in clearance with doses above 600 mcg/day is not defined.
In healthy volunteers the distribution of octreotide from plasma was rapid (t(alpha)1/2 = 0.2 h), the volume of distribution (Vdss) was estimated to be 13.6 L, and the total body clearance was 10 L/hr.
In blood, the distribution into the erythrocytes was found to be negligible and about 65% was bound in the plasma in a concentration-independent manner. Binding was mainly to lipoprotein and, to a lesser extent, to albumin.
The elimination of octreotide from plasma had an apparent half-life of 1.7 hours compared with 1-3 minutes with the natural hormone. The duration of action of Sandostatin® (octreotide acetate) is variable but extends up to 12 hours depending upon the type of tumor. About 32% of the dose is excreted unchanged into the urine. In an elderly population, dose adjustments may be necessary due to a significant increase in the half-life (46%) and a significant decrease in the clearance (26%) of the drug.
In patients with acromegaly, the pharmacokinetics differ somewhat from those in healthy volunteers. A mean peak concentration of 2.8 ng/mL (100 mcg dose) was reached in 0.7 hours after subcutaneous dosing. The volume of distribution (Vdss) was estimated to be 21.6 ± 8.5 L and the total body clearance was increased to 18 L/h. The mean percent of the drug bound was 41.2%. The disposition and elimination half-lives were similar to normals.
In patients with severe renal failure requiring dialysis, clearance was reduced to about half that found in normal subjects (from approximately 10 L/h to 4.5 L/h). The effect of hepatic diseases on the disposition of octreotide is unknown.
Sandostatin® (octreotide acetate) is indicated to reduce blood levels of growth hormone and IGF-I (somatomedin C) in acromegaly patients who have had inadequate response to or cannot be treated with surgical resection, pituitary irradiation, and bromocriptine mesylate at maximally tolerated doses. The goal is to achieve normalization of growth hormone and IGF-I (somatomedin C) levels (See DOSAGE AND ADMINISTRATION ). In patients with acromegaly, Sandostatin® (octreotide acetate) reduces growth hormone to within normal ranges in 50% of patients and reduces IGF-I (somatomedin C) to within normal ranges in 50%-60% of patients. Since the effects of pituitary irradiation may not become maximal for several years, adjunctive therapy with Sandostatin® (octreotide acetate) to reduce blood levels of growth hormone and IGF-I (somatomedin C) offers potential benefit before the effects of irradiation are manifested.
Improvement in clinical signs and symptoms or reduction in tumor size or rate of growth were not shown in clinical trials performed with Sandostatin® (octreotide acetate); these trials were not optimally designed to detect such effects.
Sandostatin® (octreotide acetate) is indicated for the symptomatic treatment of patients with metastatic carcinoid tumors where it suppresses or inhibits the severe diarrhea and flushing episodes associated with the disease.
Sandostatin® (octreotide acetate) studies were not designed to show an effect on the size, rate of growth or development of metastases.
Sandostatin® (octreotide acetate) is indicated for the treatment of the profuse watery diarrhea associated with VIP-secreting tumors. Sandostatin® (octreotide acetate) studies were not designed to show an effect on the size, rate of growth or development of metastases.
Sensitivity to this drug or any of its components.
Single doses of Sandostatin® (octreotide acetate) have been shown to inhibit gallbladder contractility and decrease bile secretion in normal volunteers. In clinical trials (primarily patients with acromegaly or psoriasis), the incidence of biliary tract abnormalities was 63% (27% gallstones, 24% sludge without stones, 12% biliary duct dilatation). The incidence of stones or sludge in patients who received Sandostatin® (octreotide acetate) for 12 months or longer was 52%. Less than 2% of patients treated with Sandostatin® (octreotide acetate) for 1 month or less developed gallstones. The incidence of gallstones did not appear related to age, sex or dose. Like patients without gallbladder abnormalities, the majority of patients developing gallbladder abnormalities on ultrasound had gastrointestinal symptoms. The symptoms were not specific for gallbladder disease. A few patients developed acute cholecystitis, ascending cholangitis, biliary obstruction, cholestatic hepatitis, or pancreatitis during Sandostatin® (octreotide acetate) therapy or following its withdrawal. One patient developed ascending cholangitis during Sandostatin® (octreotide acetate) therapy and died.
Sandostatin® (octreotide acetate) alters the balance between the counter-regulatory hormones, insulin, glucagon and growth hormone, which may result in hypoglycemia or hyperglycemia. Sandostatin® (octreotide acetate) also suppresses secretion of thyroid stimulating hormone, which may result in hypothyroidism. Cardiac conduction abnormalities have also occurred during treatment with Sandostatin® (octreotide acetate). However, the incidence of these adverse events during long-term therapy was determined vigorously only in acromegaly patients who, due to their underlying disease and/or the subsequent treatment they receive, are at an increased risk for the development of diabetes mellitus, hypothyroidism, and cardiovascular disease. Although the degree to which these abnormalities are related to Sandostatin® (octreotide acetate) therapy is not clear, new abnormalities of glycemic control, thyroid function and ECG developed during Sandostatin® (octreotide acetate) therapy as described below.
The hypoglycemia or hyperglycemia which occurs during Sandostatin® (octreotide acetate) therapy is usually mild, but may result in overt diabetes mellitus or necessitate dose changes in insulin or other hypoglycemic agents. Hypoglycemia and hyperglycemia occurred on Sandostatin® (octreotide acetate) in 3% and 16% of acromegalic patients, respectively. Severe hyperglycemia, subsequent pneumonia, and death following initiation of Sandostatin® (octreotide acetate) therapy was reported in one patient with no history of hyperglycemia.
In acromegalic patients, 12% developed biochemical hypothyroidism only, 8% developed goiter, and 4% required initiation of thyroid replacement therapy while receiving Sandostatin® (octreotide acetate). Baseline and periodic assessment of thyroid function (TSH, total and/or free T 4 ) is recommended during chronic therapy.
In acromegalics, bradycardia (<50 bpm) developed in 25%; conduction abnormalities occurred in 10% and arrhythmias occurred in 9% of patients during Sandostatin® (octreotide acetate) therapy. Other EKG changes observed included QT prolongation, axis shifts, early repolarization, low voltage, R/S transition, and early R wave progression. These ECG changes are not uncommon in acromegalic patients. Dose adjustments in drugs such as beta-blockers that have bradycardia effects may be necessary. In one acromegalic patient with severe congestive heart failure, initiation of Sandostatin® (octreotide acetate) therapy resulted in worsening of CHF with improvement when drug was discontinued. Confirmation of a drug effect was obtained with a positive rechallenge.
Several cases of pancreatitis have been reported in patients receiving Sandostatin® (octreotide acetate) therapy.
Sandostatin® (octreotide acetate) may alter absorption of dietary fats in some patients.
In patients with severe renal failure requiring dialysis, the half-life of Sandostatin® (octreotide acetate) may be increased, necessitating adjustment of the maintenance dosage.
Depressed vitamin B 12 levels and abnormal Schilling' tests have been observed in some patients receiving Sandostatin® (octreotide acetate) therapy, and monitoring of vitamin B 12 levels is recommended during chronic Sandostatin® (octreotide acetate) therapy.
Careful instruction in sterile subcutaneous injection technique should be given to the patients and to other persons who may administer Sandostatin® (octreotide acetate) Injection.
Laboratory tests that may be helpful as biochemical markers in determining and following patient response depend on the specific tumor. Based on diagnosis, measurement of the following substances may be useful in monitoring the progress of therapy:
|
Growth Hormone, IGF-I (somatomedin C)
Responsiveness to Sandostatin® (octreotide acetate) may be evaluated by determining growth hormone levels at 1-4 hour intervals for 8-12 hours post dose. Alternatively, a single measurement of IGF-I (somatomedin C) level may be made two weeks after drug initiation or dosage change. |
|
VIP (plasma vasoactive intestinal peptide)
|
Baseline and periodic total and/or free T 4 measurements should be performed during chronic therapy (see PRECAUTIONS -- General ).
Sandostatin® (octreotide acetate) has been associated with alterations in nutrient absorption, so it may have an effect on absorption of orally administered drugs. Concomitant administration of Sandostatin® (octreotide acetate) with cyclosporine may decrease blood levels of cyclosporine and result in transplant rejection.
Patients receiving insulin, oral hypoglycemic agents, beta blockers, calcium channel blockers, or agents to control fluid and electrolyte balance, may require dose adjustments of these therapeutic agents.
No known interference exists with clinical laboratory tests, including amine or peptide determinations.
Studies in laboratory animals have demonstrated no mutagenic potential of Sandostatin® (octreotide acetate).
No carcinogenic potential was demonstrated in mice treated subcutaneously for 85-99 weeks at doses up to 2000 mcg/kg/day (8x the human exposure based on body surface area). In a 116-week subcutaneous study in rats, a 27% and 12% incidence of injection site sarcomas or squamous cell carcinomas was observed in males and females, respectively, at the highest dose level of 1250 mcg/kg/day (10x the human exposure based on body surface area) compared to an incidence of 8%-10% in the vehicle control groups. The increased incidence of injection site tumors was most probably caused by irritation and the high sensitivity of the rat to repeated subcutaneous injections at the same site. Rotating injection sites would prevent chronic irritation in humans. There have been no reports of injection site tumors in patients treated with Sandostatin® (octreotide acetate) for up to 5 years. There was also a 15% incidence of uterine adenocarcinomas in the 1250 mcg/kg/day females compared to 7% in the saline control females and 0% in the vehicle control females. The presence of endometritis coupled with the absence of corpora lutea, the reduction in mammary fibroadenomas, and the presence of uterine dilatation suggest that the uterine tumors were associated with estrogen dominance in the aged female rats which does not occur in humans.
Sandostatin® (octreotide acetate) did not impair fertility in rats at doses up to 1000 mcg/kg/day, which represents 7x the human exposure based on body surface area.
Reproduction studies have been performed in rats and rabbits at doses up to 16 times the highest human dose based on body surface area and have revealed no evidence of impaired fertility or harm to the fetus due to Sandostatin® (octreotide acetate). There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in milk, caution should be exercised when Sandostatin® (octreotide acetate) is administered to a nursing woman.
Experience with Sandostatin® (octreotide acetate) in the pediatric population is limited. The youngest patient to receive the drug was 1 month old. Doses of 1-10 mcg/kg body weight were well tolerated in the young patients. A single case of an infant (nesidioblastosis) was complicated by a seizure thought to be independent of Sandostatin® (octreotide acetate) therapy.
Gallbladder abnormalities, especially stones and/or biliary sludge, frequently develop in patients on chronic Sandostatin® (octreotide acetate) therapy (See ).
In acromegalics, sinus bradycardia (<50 bpm) developed in 25%; conduction abnormalities occurred in 10% and arrhythmias developed in 9% of patients during Sandostatin® (octreotide acetate) therapy (see PRECAUTIONS -- General ).
Diarrhea, loose stools, nausea and abdominal discomfort were each seen in 34%-61% of acromegalic patients in US studies although only 2.6% of the patients discontinued therapy due to these symptoms. These symptoms were seen in 5%-10% of patients with other disorders.
The frequency of these symptoms was not dose-related, but diarrhea and abdominal discomfort generally resolved more quickly in patients treated with 300 mcg/day than in those treated with 750 mcg/day. Vomiting, flatulence, abnormal stools, abdominal distention, and constipation were each seen in less than 10% of patients.
Hypoglycemia and hyperglycemia occurred in 3% and 16% of acromegalic patients, respectively, but only in about 1.5% of other patients. Symptoms of hypoglycemia were noted in approximately 2% of patients.
In acromegalics, biochemical hypothyroidism alone occurred in 12% while goiter occurred in 6% during Sandostatin® (octreotide acetate) therapy (See PRECAUTIONS -- General ). In patients without acromegaly, hypothyroidism has only been reported in several isolated patients and goiter has not been reported.
Pain on injection was reported in 7.7%, headache in 6% and dizziness in 5%. Pancreatitis was also observed ( See and PRECAUTIONS ).
Other events (relationship to drug not established), each observed in 1%-4% of patients, included fatigue, weakness, pruritus, joint pain, backache, urinary tract infection, cold symptoms, flu symptoms, injection site hematoma, bruise, edema, flushing, blurred vision, pollakiuria, fat malabsorption, hair loss, visual disturbance and depression.
Events reported in less than 1% of patients and for which relationship to drug is not established are listed: Gastrointestinal hepatitis, jaundice, increase in liver enzymes, GI bleeding, hemorrhoids, appendicitis, gastric/peptic ulcer, gallbladder polyp; Integumentary: rash, cellulitis, petechiae, urticaria, basal cell carcinoma; Musculoskeletal: arthritis, joint effusion, muscle pain, Raynaud' phenomenon; Cardiovascular: chest pain, shortness of breath, thrombophlebitis, ischemia, congestive heart failure, hypertension, hypertensive reaction, palpitations, orthostatic BP decrease, tachycardia; CNS: anxiety, libido decrease, syncope, tremor, seizure, vertigo, Bell' Palsy, paranoia, pituitary apoplexy, increased intraocular pressure, amnesia, hearing loss, neuritis; Respiratory: pneumonia, pulmonary nodule, status asthmaticus; Endocrine galactorrhea, hypoadrenalism, diabetes insipidus, gynecomastia, amenorrhea, polymenorrhea, oligomenorrhea, vaginitis; Urogenital: nephrolithiasis, hematuria; Hematologic: anemia, iron deficiency, epistaxis; Miscellaneous: otitis, allergic reaction, increased CK, weight loss.
Evaluation of 20 patients treated for at least 6 months has failed to demonstrate titers of antibodies exceeding background levels. However, antibody titers to Sandostatin® (octreotide acetate) were subsequently reported in three patients and resulted in prolonged duration of drug action in two patients. Anaphylactoid reactions, including anaphylactic shock, have been reported in several patients receiving Sandostatin® (octreotide acetate).
No frank overdose has occurred in any patient to date. Intravenous bolus doses of 1 mg (1000 mcg) given to healthy volunteers and of 30 mg (30,000 mcg) IV over 20 minutes and of 120 mg (120,000 mcg) IV over 8 hours to research patients have not resulted in serious ill effects.
Up-to-date information about the treatment of overdose can often be obtained from a certified Regional Poison Control Center. Telephone numbers of certified Regional Poison Control Centers are listed in the Physicians' Desk Reference®.*
Mortality occurred in mice and rats given 72 mg/kg and 18 mg/kg IV, respectively.
There is no indication that Sandostatin® (octreotide acetate) has potential for drug abuse or dependence. Sandostatin® (octreotide acetate) levels in the central nervous system are negligible, even after doses up to 30,000 mcg.
Sandostatin® (octreotide acetate) may be administered subcutaneously or intravenously. Subcutaneous injection is the usual route of administration of Sandostatin® (octreotide acetate) for control of symptoms. Pain with subcutaneous administration may be reduced by using the smallest volume that will deliver the desired dose. Multiple subcutaneous injections at the same site within short periods of time should be avoided. Sites should be rotated in a systematic manner.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. Do not use if particulates and/or discoloration are observed. Proper sterile technique should be used in the preparation of parenteral admixtures to minimize the possibility of microbial contamination. Sandostatin® (octreotide acetate) is not compatible in Total Parenteral Nutrition (TPN) solutions because of the formation of a glycosyl octreotide conjugate which may decrease the efficacy of the product.
Sandostatin® (octreotide acetate) is stable in sterile isotonic saline solutions or sterile solutions of dextrose 5% in water for 24 hours. It may be diluted in volumes of 50-200 mL and infused intravenously over 15-30 minutes or administered by IV push over 3 minutes. In emergency situations (e.g.: carcinoid crisis) it may be given by rapid bolus.
The initial dosage is usually 50 mcg administered twice or three times daily. Upward dose titration is frequently required. Dosage information for patients with specific tumors follows.
Dosage may be initiated at 50 mcg t.i.d. Beginning with this low dose may permit adaptation to adverse gastrointestinal effects for patients who will require higher doses. IGF-I (somatomedin C) levels every 2 weeks can be used to guide titration. Alternatively, multiple growth hormone levels at 0-8 hours after Sandostatin® (octreotide acetate) administration permit more rapid titration of dose. The goal is to achieve growth hormone levels less than 5 ng/mL or IGF-I (somatomedin C) levels less than 1.9 U/mL in males and less than 2.2 U/mL in females. The dose most commonly found to be effective is 100 mcg t.i.d., but some patients require up to 500 mcg t.i.d. for maximum effectiveness. Doses greater than 300 mcg/day seldom result in additional biochemical benefit, and if an increase in dose fails to provide additional benefit, the dose should be reduced. IGF-I (somatomedin C) or growth hormone levels should be reevaluated at 6 month intervals.
Sandostatin® (octreotide acetate) should be withdrawn yearly for approximately 4 weeks from patients who have received irradiation to assess disease activity. If growth hormone or IGF-I (somatomedin C) levels increase and signs and symptoms recur, Sandostatin® (octreotide acetate) therapy may be resumed.
The suggested daily dosage of Sandostatin® (octreotide acetate) during the first 2 weeks of therapy ranges from 100-600 mcg/day in 2-4 divided doses (mean daily dosage is 300 mcg). In the clinical studies, the median daily maintenance dosage was approximately 450 mcg, but clinical and biochemical benefits were obtained in some patients with as little as 50 mcg, while others required doses up to 1500 mcg/day. However, experience with doses above 750 mcg/day is limited.
Daily dosages of 200-300 mcg in 2-4 divided doses are recommended during the initial 2 weeks of therapy (range 150-750 mcg) to control symptoms of the disease. On an individual basis, dosage may be adjusted to achieve a therapeutic response, but usually doses above 450 mcg/day are not required.
Sandostatin® (octreotide acetate) Injection is available in 1 mL ampuls and 5 mL multi-dose vials as follows:
50 mcg/mL octreotide (as acetate)
Package of 20 ampuls (NDC 0078-0180-03)
100 mcg/mL octreotide (as acetate)
Package of 20 ampuls (NDC 0078-0181-03)
500 mcg/mL octreotide (as acetate)
Package of 20 ampuls (NDC 0078-0182-03)
200 mcg/mL octreotide (as acetate)
Box of one (NDC 0078-0183-25)
1000 mcg/mL octreotide (as acetate)
Box of one (NDC 0078-0184-25)
For prolonged storage, Sandostatin® (octreotide acetate) ampuls and multi-dose vials should be stored at refrigerated temperatures 2°-8°C (36°-46°F) and protected from light. At room temperature, (20°-30°C or 70°-86°F), Sandostatin® (octreotide acetate) is stable for 14 days if protected from light. The solution can be allowed to come to room temperature prior to administration. Do not warm artificially. After initial use, multiple dose vials should be discarded within 14 days. Ampuls should be opened just prior to administration and the unused portion discarded.
The ampuls and multi-dose vials are manufactured by: NOVARTIS PHARMA AG, Basle, Switzerland
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936
*Medical Economics Company, Inc.
© 1999 Novartis
REV: MAY 1999 T1999-40
 |