 |
WARNINGVUMON® (teniposide injection) is a cytotoxic drug, which should be administered under the supervision of a qualified physician experienced in the use of cancer chemotherapeutic agents. Appropriate management of therapy and complications is possible only when adequate treatment facilities are readily available. Severe myelosuppression with resulting infection or bleeding may occur. Hypersensitivity reactions, including anaphylaxis-like symptoms, may occur with initial dosing or at repeated exposure to VUMON. Epinephrine, with or without corticosteroids and antihistamines has been employed to alleviate hypersensitivity reaction symptoms. |
VUMON® (teniposide injection) (also commonly known as VM-26), is supplied as a sterile nonpyrogenic solution in a nonaqueous medium intended for dilution with a suitable parenteral vehicle prior to intravenous infusion. VUMON is available in 50 mg (5 mL) ampules. Each mL contains 10 mg teniposide, 30 mg benzyl alcohol, 60 mg N,N-dimethylacetamide, 500 mg purified Cremophor® EL (polyoxyethylated castor oil) * and 42.7 percent (V/V) dehydrated alcohol. The pH of the clear solution is adjusted to approximately 5 with maleic acid.
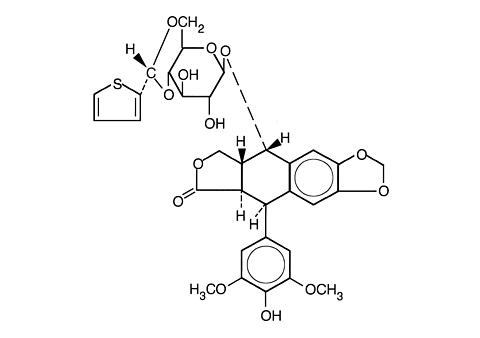
Teniposide is a semisynthetic derivative of podophyllotoxin. The chemical name for teniposide is 4'-demethylepipodophyllotoxin 9-[4,6-0-(R)-2- thenylidene-(beta)-D-glucopyranoside]. Teniposide differs from etoposide, another podophyllotoxin derivative, by the substitution of a thenylidene group on the glucopyranoside ring.
Teniposide has the following structural formula:
|
|
Teniposide is a white to off-white crystalline powder with the empirical formula C 32 H 32 O 13 S and a molecular weight of 656.66. It is a lipophilic compound with a partition coefficient value (octanol/water) of approximately 100. Teniposide is insoluble in water and ether. It is slightly soluble in methanol and very soluble in acetone and dimethylformamide.
Teniposide is a phase-specific cytotoxic drug, acting in the late S or early G 2 phase of the cell cycle, thus preventing cells from entering mitosis.
Teniposide causes dose-dependent single- and double-stranded breaks in DNA and DNA: protein cross-links. The mechanism of action appears to be related to the inhibition of type II topoisomerase activity since teniposide does not intercalate into DNA or bind strongly to DNA. The cytotoxic effects of teniposide are related to the relative number of double-stranded DNA breaks produced in cells, which are a reflection of the stabilization of a topoisomerase II-DNA intermediate.
Teniposide has a broad spectrum of in vivo antitumor activity against murine tumors, including hematologic malignancies and various solid tumors. Notably, teniposide is active against sublines of certain murine leukemias with acquired resistance to cisplatin, doxorubicin, amsacrine, daunorubicin, mitoxantrone or vincristine.
Plasma drug levels declined biexponentially following intravenous infusion (155 mg/m 2 over 1 to 2.5 hours) of VUMON given to eight children (4-11 years old) with newly diagnosed acute lymphoblastic leukemia (ALL). The observed average pharmacokinetic parameters and associated coefficients of variation (CV%) based on a two-compartmental model analysis of the data are as follows:
|
There appears to be some association between an increase in serum alkaline phosphatase or gamma glutamyl-transpeptidase and a decrease in plasma clearance of teniposide. Therefore, caution should be exercised if VUMON is to be administered to patients with hepatic dysfunction.
In adults, at doses of 100 to 333 mg/m 2 /day, plasma levels increased linearly with dose. Drug accumulation in adult patients did not occur after daily administration of VUMON for 3 days. In pediatric patients, maximum plasma concentrations (C max ) after infusions of 137 to 203 mg/m 2 over a period of one to two hours exceeded 40 µg/mL; by 20 to 24 hours after infusion plasma levels were generally < 2µg/mL.
Renal clearance of parent teniposide accounts for about 10 percent of total body clearance. In adults, after intravenous administration of 10 mg/kg or 67 mg/m 2 of tritium-labeled teniposide, 44 percent of the radiolabel was recovered in urine (parent drug and metabolites) within 120 hours after dosing. From 4 to 12 percent of a dose is excreted in urine as parent drug. Fecal excretion of radioactivity within 72 hours after dosing accounted for 0 to 10 percent of the dose.
Mean steady-state volumes of distribution range from 8 to 44 L/m 2 for adults and 3 to 11 L/m 2 for children. The blood-brain barrier appears to limit diffusion of teniposide into the brain, although in a study in patients with brain tumors, CSF levels of teniposide were higher than CSF levels reported in other studies of patients who did not have brain tumors.
Teniposide is highly protein bound. In vitro plasma protein binding of teniposide is >99 percent. The high affinity of teniposide for plasma proteins may be an important factor in limiting distribution of drug within the body. Steady state volume of distribution of the drug increases with a decrease in plasma albumin levels. Therefore, careful monitoring of children with hypoalbuminemia is indicated during therapy. Levels of teniposide in saliva, CSF and malignant ascites fluid are low relative to simultaneously measured plasma levels.
The pharmacokinetic characteristics of teniposide differ from those of etoposide, another podophyllotoxin. Teniposide is more extensively bound to plasma proteins, and its cellular uptake is greater. Teniposide also has a lower systemic clearance, a longer elimination half-life, and is excreted in the urine as parent drug to a lesser extent than etoposide.
In a study at St. Jude Children's Research Hospital (SJCRH), 9 children with acute lymphocytic leukemia (ALL) failing induction therapy with a cytarabine-containing regimen, were treated with VUMON (teniposide injection) plus cytarabine. Three of these patients were induced into complete remission with durations of remission of 30 weeks, 59 weeks, and 13 years. In another study at SJCRH, 16 children with ALL refractory to vincristine/prednisone-containing regimens were treated with VUMON plus vincristine and prednisone. Three of these patients were induced into complete remission with durations of remission of 5.5, 37, and 73 weeks. In these two studies patients served as their own control based on the premise that long term complete remissions could not be achieved by re-treatment with drugs to which they had previously failed to respond.
VUMON, in combination with other approved anticancer agents, is indicated for induction therapy in patients with refractory childhood acute lymphoblastic leukemia.
VUMON is generally contraindicated in patients who have demonstrated a previous hypersensitivity to teniposide and/or Cremophor® EL (polyoxyethylated castor oil).
VUMON is a potent drug and should be used only by physicians experienced in the administration of cancer chemotherapeutic drugs. Blood counts as well as renal and hepatic function tests should be carefully monitored prior to and during therapy.
Patients being treated with VUMON should be observed frequently for myelosuppression both during and after therapy. Dose-limiting bone marrow suppression is the most significant toxicity associated with VUMON therapy. Therefore, the following studies should be obtained at the start of therapy and prior to each subsequent dose of VUMON: hemoglobin, white blood cell count and differential and platelet count. If necessary, repeat bone marrow examination should be performed prior to the decision to continue therapy in the setting of severe myelosuppression.
Physicians should be aware of the possible occurrence of a hypersensitivity reaction variably manifested by chills, fever, urticaria, tachycardia, bronchospasm, dyspnea, hypertension or hypotension and facial flushing. This reaction may occur with the first dose of VUMON and may be life threatening if not treated promptly with antihistamines, corticosteroids, epinephrine, intravenous fluids and other supportive measures as clinically indicated. The exact cause of these reactions is unknown. They may be due to the Cremophor® EL (polyoxyethylated castor oil) component of the vehicle or to teniposide itself. 1 Patients who have experienced prior hypersensitivity reactions to VUMON are at risk for recurrence of symptoms and should only be re-treated with VUMON if the antileukemic benefit already demonstrated clearly outweighs the risk of a probable hypersensitivity reaction for that patient. When a decision is made to re-treat a patient with VUMON in spite of an earlier hypersensitivity reaction, the patient should be pretreated with corticosteroids and antihistamines and receive careful clinical observation during and after VUMON infusion. In the clinical experience with VUMON at SJCRH and the National Cancer Institute (NCI), re-treatment of patients with prior hypersensitivity reactions has been accomplished using measures described above. To date, there is no evidence to suggest cross-sensitization between VUMON and VePesid® (etoposide).
One episode of sudden death, attributed to probable arrhythmia and intractable hypotension has been reported in an elderly patient receiving VUMON combination therapy for a non-leukemic malignancy. (See ADVERSE REACTIONS .) Patients receiving VUMON treatment should be under continuous observation for at least the first 60 minutes following the start of the infusion and at frequent intervals thereafter. If symptoms or signs of anaphylaxis occur, the infusion should be stopped immediately, followed by the administration of epinephrine, corticosteroids, antihistamines, pressor agents, or volume expanders at the discretion of the physician. An aqueous solution of epinephrine 1:1000 and a source of oxygen should be available at the bedside.
For parenteral administration, VUMON should be given only by slow intravenous infusion (lasting at least 30- to 60-minutes) since hypotension has been reported as a possible side effect of rapid intravenous injection, perhaps due to a direct effect of Cremophor® EL. 2,3 If clinically significant hypotension develops, the VUMON infusion should be discontinued. The blood pressure usually normalizes within hours in response to cessation of the infusion and administration of fluids or other supportive therapy as appropriate. If the infusion is restarted, a slower administration rate should be used and the patient should be carefully monitored.
Acute central nervous system depression and hypotension have been observed in patients receiving investigational infusions of high-dose VUMON who were pretreated with antiemetic drugs. The depressant effects of the antiemetic agents and the alcohol content of the VUMON formulation may place patients receiving higher than recommended doses of VUMON at risk for central nervous system depression.
VUMON may cause fetal harm when administered to a pregnant woman. VUMON has been shown to be teratogenic and embryotoxic in laboratory animals. In pregnant rats intravenous administration of VUMON, 0.1-3 mg/kg (0.6-18 mg/m 2 ), every second day from day 6 to day 16 post coitum caused dose-related embryotoxicity and teratogenicity. Major anomalies included spinal and rib defects, deformed extremities, anophthalmia and celosomia.
There are no adequate and well-controlled studies in pregnant women. If VUMON is used during pregnancy, or if the patient becomes pregnant while receiving this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant during therapy with VUMON.
General: In all instances where the use of VUMON is considered for chemotherapy, the physician must evaluate the need and usefulness of the drug against the risk of adverse reactions. Most such adverse reactions are reversible if detected early. If severe reactions occur, the drug should be reduced in dosage or discontinued and appropriate corrective measures should be taken according to the clinical judgment of the physician. Reinstitution of VUMON therapy should be carried out with caution, and with adequate consideration of the further need for the drug and alertness as to possible recurrence of toxicity.
VUMON must be administered as an intravenous infusion. Care should be taken to ensure that the intravenous catheter or needle is in the proper position and functional prior to infusion. Improper administration of VUMON may result in extravasation causing local tissue necrosis and/or thrombophlebitis. In some instances, occlusion of central venous access devices has occurred during 24-hour infusion of VUMON at a concentration of 0.1 to 0.2 mg/mL. Frequent observation during these infusions is necessary to minimize this risk. 4,5
Laboratory Tests: Periodic complete blood counts and assessments of renal and hepatic function should be done during the course of VUMON treatment. They should be performed prior to therapy and at clinically appropriate intervals during and after therapy. There should be at least one determination of hematologic status prior to therapy with VUMON.
Drug Interactions: In a study in which 34 different drugs were tested, therapeutically relevant concentrations of tolbutamide, sodium salicylate and sulfamethizole displaced protein-bound teniposide in fresh human serum to a small but significant extent. Because of the extremely high binding of teniposide to plasma proteins, these small decreases in binding could cause substantial increases in free drug levels in plasma which could result in potentiation of drug toxicity. Therefore, caution should be used in administering VUMON (teniposide injection) to patients receiving these other agents. There was no change in the plasma kinetics of teniposide when coadministered with methotrexate. However, the plasma clearance of methotrexate was slightly increased. An increase in intracellular levels of methotrexate was observed in vitro in the presence of teniposide.
Carcinogenesis, Mutagenesis, Impairment of Fertility: Children at SJCRH with ALL in remission who received maintenance therapy with VUMON at weekly or twice weekly doses (plus other chemotherapeutic agents), had a relative risk of developing secondary acute nonlymphocytic leukemia (ANLL) approximately 12 times that of patients treated according to other less intensive schedules. 6
A short course of VUMON for remission-induction and/or consolidation therapy was not associated with an increased risk of secondary ANLL, but the number of patients assessed was small. The potential benefit from VUMON must be weighed on a case by case basis against the potential risk of the induction of a secondary leukemia. The carcinogenicity of teniposide has not been studied in laboratory animals. Compounds with similar mechanisms of action and mutagenicity profiles have been reported to be carcinogenic and teniposide should be considered a potential carcinogen in humans. Teniposide has been shown to be mutagenic in various bacterial and mammalian genetic toxicity tests. These include positive mutagenic effects in the Ames/Salmonella and B. subtilis bacterial mutagenicity assays. Teniposide caused gene mutations in both Chinese hamster ovary cells and mouse lymphoma cells and DNA damage as measured by alkaline elution in human lung carcinoma derived cell lines. In addition, teniposide induced aberrations in chromosome structure in primary cultures of human lymphocytes in vitro and in L5178y/TK +/- mouse lymphoma cells in vitro . Chromosome aberrations were observed in vivo in the embryonic tissue of pregnant Swiss albino mice treated with teniposide. Teniposide also caused a dose-related increase in sister chromatid exchanges in Chinese hamster ovary cells and it has been shown to be embryotoxic and teratogenic in rats receiving teniposide during organogenesis. Treatment of pregnant rats I.V. with doses between 1.0 and 3.0 mg/kg/day on alternate days from day 6 to 16 post coitum caused retardation of embryonic development, prenatal mortality and fetal abnormalities.
Pregnancy Pregnancy "Category D". (See .)
Nursing Mothers: It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of VUMON therapy to the mother.
Patients with Down' Syndrome: Patients with both Down' Syndrome and leukemia may be especially sensitive to myelosuppressive chemotherapy, therefore, initial dosing with VUMON should be reduced in these patients. It is suggested that the first course of VUMON should be given at half the usual dose. Subsequent courses may be administered at higher dosages depending on the degree of myelosuppression and mucositis encountered in earlier courses in an individual patient.
The table below presents the incidences of adverse reactions derived from an analysis of data contained within literature reports of 7 studies involving 303 pediatric patients in which VUMON was administered by injection as a single agent in a variety of doses and schedules for a variety of hematologic malignancies and solid tumors. The total number of patients evaluable for a given event was not 303 since the individual studies did not address the occurrence of each event listed. Five of these seven studies assessed VUMON activity in hematologic malignancies, such as leukemia. Thus, many of these patients had abnormal hematologic status at start of therapy with VUMON and were expected to develop significant myelosuppression as an endpoint of treatment.
|
||||||||||||||||||||||||||||||||||||||||||||
Hematologic Toxicity: VUMON, when used with other chemotherapeutic agents for the treatment of ALL, results in severe myelosuppression. Early onset of profound myelosuppression with delayed recovery can be expected when using the doses and schedules of VUMON necessary for treatment of refractory ALL, since bone marrow hypoplasia is a desired endpoint of therapy. The occurrence of acute non-lymphocytic leukemia (ANLL), with or without a preleukemic phase, has been reported in patients treated with VUMON in combination with other antineoplastic agents. See PRECAUTIONS , Carcinogenesis, Mutagenesis, Impairment of Fertility.
Gastrointestinal Toxicity: Nausea and vomiting are the most common gastrointestinal toxicities, having occurred in 29 percent of evaluable pediatric patients. The severity of this nausea and vomiting is generally mild to moderate.
Hypotension Transient hypotension following rapid intravenous administration has been reported in 2 percent of evaluable pediatric patients. One episode of sudden death, attributed to probable arrhythmia and intractable hypotension, has been reported in an elderly patient receiving VUMON combination therapy for a non-leukemic malignancy.
No other cardiac toxicity or electrocardiographic changes have been documented. No delayed hypotension has been noted.
Allergic Reactions: Hypersensitivity reactions characterized by chills, fever, tachycardia, flushing, bronchospasm, dyspnea, and blood pressure changes (hypertension or hypotension) have been reported to occur in approximately 5 percent of evaluable pediatric patients receiving intravenous VUMON. The incidence of hypersensitivity reactions to VUMON appears to be increased in patients with brain tumors, and in patients with neuroblastoma. 1
Central Nervous System: Acute central nervous system depression and hypotension have been observed in patients receiving investigational infusions of high-dose VUMON (teniposide injection) who were pretreated with antiemetic drugs. The depressant effects of the antiemetic agents and the alcohol content of the VUMON formulation may place patients receiving higher than recommended doses of VUMON at risk for central nervous system depression.
Alopecia: Alopecia, sometimes progressing to total baldness, was observed in 9 percent of evaluable pediatric patients who received VUMON as single agent therapy. It was usually reversible.
There is no known antidote for VUMON overdosage. The anticipated complications of overdosage are secondary to bone marrow suppression. Treatment should consist of supportive care including blood products and antibiotics as indicated.
NOTE : Contact of undiluted VUMON with plastic equipment or devices used to prepare solutions for infusion may result in softening or cracking and possible drug product leakage. This effect has not been reported with diluted solutions of VUMON.
In order to prevent extraction of the plasticizer DEHP [di(2-ethylhexyl)phtalate], solutions of VUMON should be prepared in non-DEHP containing LVP containers such as glass or polyolefin plastic bags or containers.
VUMON solutions should be administered with non-DEHP containing I.V. administration sets.
In one study, childhood ALL patients failing induction therapy with a cytarabine-containing regimen were treated with the combination of VUMON 165 mg/m 2 and cytarabine 300 mg/m 2 intravenously, twice weekly for 8-9 doses. In another study, patients with childhood ALL refractory to vincristine/prednisone-containing regimens were treated with the combination of VUMON 250 mg/m 2 and vincristine 1.5 mg/m 2 intravenously, weekly for 4-8 weeks and prednisone 40 mg/m 2 orally × 28 days.
Adequate data in patients with hepatic insufficiency and/or renal insufficiency are lacking, but dose adjustments may be necessary for patients with significant renal or hepatic impairment.
Preparation and Administration Precautions: VUMON is a cytotoxic anticancer drug and as with other potentially toxic compounds, caution should be exercised in handling and preparing the solution of VUMON. Skin reactions associated with accidental exposure to VUMON may occur. The use of gloves is recommended. If VUMON solution contacts the skin, immediately wash the skin thoroughly with soap and water. If VUMON contacts mucous membranes, the membranes should be flushed thoroughly with water.
Preparation for Intravenous Administration: VUMON must be diluted with either 5 percent Dextrose Injection, USP or 0.9 percent Sodium Chloride Injection, USP, to give final teniposide concentrations of 0.1 mg/mL, 0.2 mg/mL, 0.4 mg/mL or 1.0 mg/mL. Solutions prepared in 5 percent Dextrose Injection, USP or 0.9 percent Sodium Chloride Injection, USP at teniposide concentrations of 0.1 mg/mL, 0.2 mg/mL or 0.4 mg/mL are stable at room temperature for up to 24 hours after preparation. VUMON solutions prepared at a final teniposide concentration of 1.0 mg/mL should be administered within 4 hours of preparation to reduce the potential for precipitation. Refrigeration of VUMON solutions is not recommended. Stability and use times are identical in glass and plastic parenteral solution containers.
Although solutions are chemically stable under the conditions indicated, precipitation of teniposide may occur at the recommended concentrations, especially if the diluted solution is subjected to more agitation than is recommended to prepare the drug solution for parenteral administration. 7 In addition, storage time prior to administration should be minimized and care should be taken to avoid contact of the diluted solution with other drugs or fluids. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit. Precipitation has been reported during 24-hour infusions of VUMON diluted to teniposide concentrations of 0.1 to 0.2 mg/mL, resulting in occlusion of central venous access catheters in several patients. 4,5 Heparin solution can cause precipitation of teniposide, therefore, the administration apparatus should be flushed thoroughly with 5 percent Dextrose Injection or 0.9 percent Sodium Chloride Injection, USP before and after administration of VUMON. 5
Hypotension has been reported following rapid intravenous administration; it is recommended that the VUMON solution be administered over at least a 30 to 60-minute period. VUMON should not be given by rapid intravenous injection.
In a 24-hour study under simulated conditions of actual use of the product relative to dilution strength, diluent and administration rates, dilutions at 0.1 to 1.0 mg/mL were chemically stable for at least 24 hours. Data collected for the presence of the extractable DEHP [di(2-ethylhexyl)phtalate] from PVC containers show that levels increased with time and concentration of the solutions. The data appeared similar for 0.9 percent Sodium Chloride Injection, USP, and 5 percent Dextrose Injection, USP. Consequently, the use of PVC containers is not recommended.
Similarly, the use of non-DEHP I.V. administration sets is recommended. Lipid administration sets or low DEHP containing nitroglycerin sets will keep patients' exposure to DEHP at low levels and are suitable for use. The diluted solutions are chemically and physically compatible with the recommended I.V. administration sets and LVP containers for up to 24 hours at ambient room temperature and lighting conditions. Because of the potential for precipitation, compatibility with other drugs, infusion materials or I.V. pumps cannot be assured.
Stability Unopened ampules of VUMON are stable until the date indicated on the package when stored under refrigeration (2°-8°C) in the original package. Freezing does not adversely affect the product.
VUMON® (teniposide injection)
NDC 0015-3075-19
50 mg/5 mL sterile clear, colorless glass ampules individually packaged in a carton.NDC 0015-3075-97
50 mg/5 mL sterile clear, colorless glass ampules individually nested in a carton tray of 10 ampules per tray.
Storage: Store the unopened ampules under refrigeration (2°-8°C). Retain in original package to protect from light.
Handling and Disposal: Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on this subject have been published. 8-14 There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate.
U.S. Patent No. 3,524,844
BRISTOL LABORATORIES®
ONCOLOGY PRODUCTS
A Bristol-Myers Squibb Company
Princeton, NJ 08543
U.S.A.
Made in Italy
K7-B001-2-00 1050966A1
Issued October 1998